
微生物细胞工厂的智能设计进展
2022-01-10 03:09张震曾雪城秦磊李春
化工学报 2021年12期
张震,曾雪城,秦磊,李春,2
(1清华大学化学工程系生物化工研究所/工业生物催化教育部重点实验室,北京 100084;2清华大学合成与系统生物学研究中心,北京 100084)
引 言
“碳达峰、碳中和”作为我国的战略发展规划,对社会各行各业的发展将产生深远影响,其中对于制造业而言,生物制造基于生物催化过程可生产各种高附加值产品,且以可再生的生物质为原料,是未来制造业可持续发展的重要方向。微生物细胞工厂通过在微生物底盘细胞中表达异源生物合成途径来生产目标化合物,是绿色生物制造的重要实现形式。当下结构已报道的天然产物超过了30万种,目前利用微生物细胞工厂实现了生物合成途径已知的青蒿酸[1]、1,3-丙二醇[2]、法尼烯[3]、甘草次酸[4]、PHA[5]、1,3-丁二醇[6]等多种高附加值化合物的生产,产品广泛应用于化工、医药、能源、食品等领域。然而,生物合成途径完全解析的天然产物只有不到3万种,尚有大量天然产物的生物合成途径未解析,严重阻碍了微生物细胞工厂的发展[7]。传统微生物细胞工厂的设计和构建方式通量小、效率低,亟需自动化的设计工具代替传统依赖经验和试错的设计构建方式,以加速未知途径化合物生物合成途径的解析及其微生物细胞工厂的合成设计。
设计-构建-测试-学习(design-build-testlearn,DBTL)循环是开发微生物细胞工厂的基本研究思路,使用递归循环的方式使设计的微生物细胞工厂逐渐提高得率、速率及产量等生产目标。设计(design)是DBTL循环的第一步,对细胞工厂能否成功构建产生重要影响。基于有机化学和生物化学的专业知识、文献报道及自身实践经验绘制潜在的生物合成途径的设计方法通常仅适用于化学结构简单的目标化合物。此外,当前微生物细胞工厂中所设计途径与合成调控元件的组合较为盲目或随机,只能逐次对元件进行优化,而且设计出的途径不能很好与底盘细胞适配。这些因素导致传统微生物细胞工厂的设计和构建通量小、效率低,且验证过程周期长,例如,阿米瑞斯生物技术公司(Amyris Biotechnologies)构建微生物细胞工厂生产抗疟疾前体青蒿素花费了十年时间、150人年的工作量,实现微生物生产法尼烯花费了四年时间、130~575人年;杜邦公司(DuPont)和杰能科公司(Genencor)分别花费约15和575来实现微生物细胞工厂生产1,3-丙二醇[8]。然而,组学时代的到来和生物数据资源的爆炸式增长已经改变了生物制造领域的研究模式,为生物制造和微生物细胞工厂设计提供了新的发展机遇。因此,在大数据基础上的智能化微生物细胞工厂设计方法成为加速微生物细胞工厂设计构建的关键。
借鉴电子设计自动化(electronic design automation,EDA,指利用计算机辅助设计软件,来完成超大规模集成电路芯片的功能设计、验证、物理设计等流程的设计方式)的概念,微生物细胞工厂的智能设计可采用生物设计自动化(biological design of automation,BDA)的形式,立足于生物数据库中的丰富资源,通过一系列算法完成细胞工厂的自动化设计,实现生物合成途径的预测与筛选、调控元件的设计、途径与元件组装设计、设计途径与底盘代谢网络适配等功能。通过生物合成途径的预测与筛选,对途径未知化合物提供可靠性较强、效率较高的候选途径方案,辅助途径设计,此外,从全局出发预测潜在的分支途径,以实现对实验中可能出现的副产物进行快速定位,进而指导细胞工厂的构建。通过调控元件的设计及元件与途径的组合设计,进一步提高预测途径与底盘菌株的适配性及可靠性,为细胞工厂后续的构建、测试和学习指明方向。
1 逆合成算法预测生物合成途径
化合物生物合成途径的设计主要采用生物逆合成算法,其思想来源于化学逆合成,分为生物逆合成途径的预测和途径筛选两个环节。在逆合成途径预测阶段,通过使用一组在原子水平上描述底物和产物分子之间化学转化模式的生化反应规则,推测合成目标化合物的反应及催化该步反应的酶,实现将输入化合物(即目标化合物)转化为一系列中间化合物,并最终转化为前体化合物的过程。生物逆合成算法按预测中间化合物的方式不同可分为两大类:一是在数据库中检索已知代谢反应并预测反应和中间化合物,所预测的中间化合物种类受到化合物数据库规模的限制,如FMM、DESHARKY和Metabolic tinker等工具;二是基于泛化的生化反应规则来预测新反应且可产生数据库中不存在的新化合物,如XTMS[9]、RetroPath[10]、RetroPath2.0[11]、RetroPath RL[12]、novoPathFinder[13]等工具(表1),这一类基于反应规则的算法在生物逆合成预测中具有更大的应用潜力,因此下述生物逆合成预测流程主要介绍基于反应规则的预测算法。
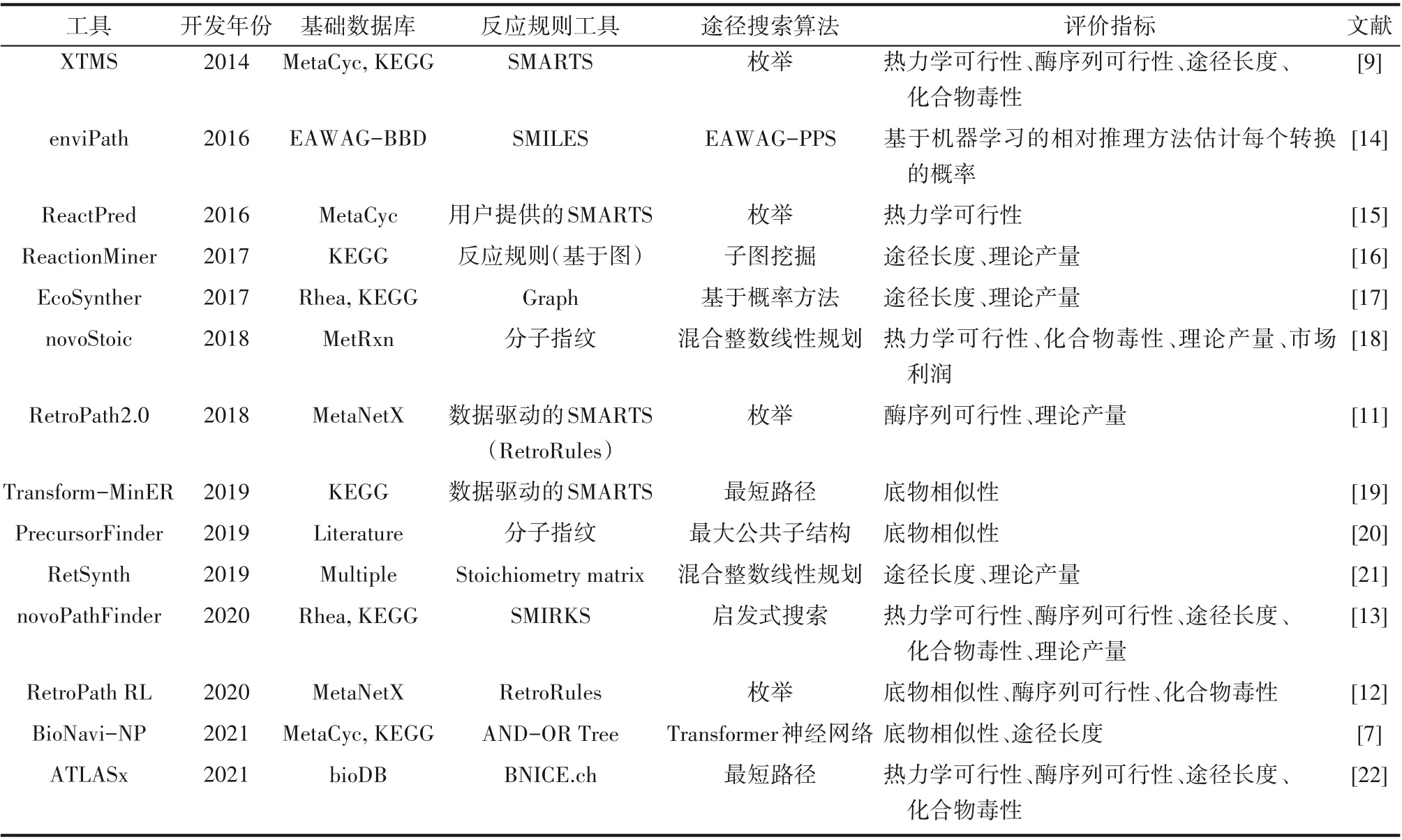
表1 生物逆合成工具Table1 Retrobiosynthesis tools
1.1 生物逆合成途径预测流程
生物合成途径由多步生化反应级联组合而成,后一步反应的前体化合物作为前一步反应的目标产物,相当于多个特殊的单步反应的组合,因此生物合成途径的预测关键在于对某一指定化合物的单步生物合成反应的预测。单步生物合成反应的预测以普遍酶具有底物杂泛性的假设条件为前提,即认为酶可以催化与底物具有相似化学结构的化合物,因此可借鉴已报道生化反应的转化模式及相应的酶序列来预测与底物具有相似结构化合物发生的新生化反应。同时存在一些计算工具可对酶的底物杂泛性进行预测,如EPP-HMCNF[23]可根据BRENDA数据库中酶-底物的互作预测可催化给定查询化合物的不同种类的酶。基于上述假设单步反应的预测流程可以概括为,对于目标化合物Q(query),在化合物数据库中检索与之结构相似的化合物M1、M2(match),在生化反应数据库中检索M1、M2的相关反应R1、R2(reaction),提取相应的反应规则及酶序列,将其应用在目标化合物Q上,从而得到目标化合物Q的生成反应、催化该步反应的酶序列及直接前体化合物Q11、Q21(图1,步骤1~5)。
通过对预测前体迭代应用单步预测反应,可逐步延伸预测途径的长度,直至达到规定的终止条件。通常以预测途径达到规定步长、预测的前体化合物为指定化合物或指定底盘宿主内源化合物等易于计算的指标作为终止条件。由于化合物相关合成反应并不单一,因此预测出的合成反应通常具有许多分支,从而使得最终的逆合成途径通常以树的形式呈现,被称为逆合成网络(图1,步骤6~7),其中目标化合物为根节点、中间化合物为子节点、指定起始化合物或底盘宿主可内源合成的化合物为叶子结点,生化反应及催化该步反应的酶序列为连接节点的边。基于逆合成网络的树形特点,一些同样呈现为树形的人工智能算法被应用到逆合成网络的生成中,如RetroPath RL[12]利用蒙特卡洛树搜索算法(Monte Carlo tree search)的选择-扩展-随机模拟-反向传播四个过程对预测途径进行延伸,最终生成逆合成网络。

图1 生物逆合成途径预测流程图Fig.1 Workflow of retrobiosynthesis
1.2 生化数据库
在生物合成途径的预测中生化数据库为预测算法的实现奠定了基础,所需要的生化数据库包括化合物数据库、生化反应数据库、代谢数据库、酶数据库及细胞模型。KEGG[24]、KNApSAcK[25]等化合物数据库及天然产物词典(Dictionary of Natural Products,DNP)(https://dnp.chemnetbase.com)和Super NaturalⅡ[26]等天然产物数据库可被用于相关反应已有文献报道的相似化合物的搜索。生化反应数据库用于提取反应规则,如ATLAS[27]数据库中收录了超过14万条反应,ATLASx[22]数据库中包含超过500万个预测反应。基于代谢途径中反应间的级联关系可将反应规则构建为反应规则网络,为长途径的预测提供可能。常用的代谢途径数据库包括:KEGG PATHWAY、MetaCyc、Reactome和UM-BBD等。酶数据库将提供催化生化反应可用的酶序列,如BRENDA是一个综合酶信息数据库,包含了酶促反应和相关的代谢通路。细胞模型为基因组规模代谢网络模型(genome-scale metabolic model,GEM),GEM模型可提供底盘宿主中存在的化合物、生化反应等信息,并可对代谢途径产量进行模拟计算、为预测途径的优化提供手段。大肠杆菌、酿酒酵母等常用模式生物均发展了系列GEM模型。目前也创建了一些综合数据库,如biochem4j图数据库中包含36765条反应信息、19735条化合物信息、245704条酶序列和8431个细胞模型。
1.3 化合物描述符
生物合成途径由一系列基于可进行酶催化转化的化合物组成,化学结构式是这些化合物的基础表示方式,可表示所有原子通过化学键与其相邻原子连接的原子键合环境信息。在生物逆合成途径预测中通常采用化学模式语言和分子指纹两种方式对化合物的结构式进行数字化编码,以满足后续提取反应规则及搜索结构相似性化合物的需求。
1.3.1 化学模式语言 在化学信息学中,SMILES(simplified molecular-input line-entry system)和SMARTS(smiles arbitrary target specification)是两种已有明确定义的化学模式语言,其中SMILES可以将化合物的二维结构式表示为ASCⅡ字符串,主要由原子和化学键两种基本符号组成;SMARTS是SMILES的延伸,允许使用通配符表示原子和化学键,如SMARTS中符号[C,N]表示该原子是碳(C)或氮(N),符号~可匹配任何化学键[28-30][图2(a)],这两种化学模式语言在生物逆合成算法中常被用于表示化学反应中反应物和产物的结构变化[9,11,13,15]。
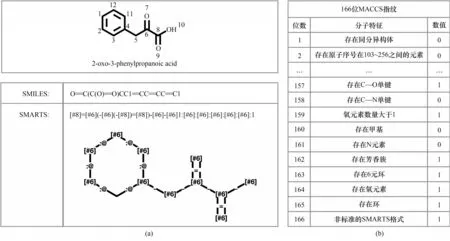
图2 化学模式语言与分子指纹Fig.2 Chemical model language and molecular fingerprints
1.3.2 分子指纹 通过预定义KEGG原子类型[31]、官能团等子结构将化合物结构分解为一个个子结构的累积,并将化合物中子结构的数量和各种物理化学特性编码为二进制变量的位串,形成化合物的分子指纹。可以使用RDKit和PaDEL-descriptor等软件包来生成分子指纹,RDkit[32]是一个用于化学信息学的开源工具包,可以生成RDKit指纹、Morgan指纹、Avalon指纹和MACCS指纹四种分子指纹,例如MACCS指纹可表示为长度为166位的向量,每一位对应一个分子特征,当化合物中存在此类特征时该位数值为1,否则为0[图2(b)]。PaDEL-descriptor[33]可 生 成PubChem指 纹、CDK指纹、CDKextend指纹、子结构指纹和GraphOnly指纹五种分子指纹。这些指纹一方面可基于Tonimoto等相似性算法用于从化合物数据库中快速搜索与查询化合物具有相似结构的化合物,另一方面可用作相似性搜索、分类和回归等各种机器学习任务的输入。
1.4 反应规则
反应规则(也称为反应描述符)为产物描述符与底物描述符之间的净差,通过底物结构式与产物结构式间的原子-原子映射比对而得到,描述了底物向产物转化时反应中心原子的键合环境变化(图3)。目前对反应规则的研究多以反应中某一个主要的反应物向相应产物的一对一转化模式为主。反应规则可作为一种模块化操作,适用于与底物结构相似的化合物上,可预测目标产物的合成反应及相应前体化合物。反应规则可以从已知反应的数据库中自动提取[34-35],也可以通过手动输入生产精简的专家反应规则集[36]。
1.4.1 基于生化反应数据库的反应规则 反应规则的自动提取需要经历(1)反应物-产物对的识别;(2)原子-原子映射;(3)反应中心原子、反应基团及保守基团的识别和(4)提取反应规则四个过程。PathPred中将每个反应中反应对(反应物-产物对)中的匹配区和非匹配区之间的边界原子分别定义为反应中心R原子、差异区域D原子和匹配区域M原子,引入了R-D-M原子模式。KEGG为反应对提供了基于原子映射的结构对齐信息,并构建了KEGG RPAIR数据库。上述反应规则表示了反应物-产物间的最大结构差异,而XTMS[9]基于SMILES化学模式语言描述化学反应,同时将原子映射编号附加到反应物和产物侧的相应原子上,以反映原子身份并跟踪反应中原子的转移;通过调整反应中心原子周围环境的大小(即直径d)可获得不同泛化水平的反应规则,当d=0时仅包括反应中心原子,d=1时包括反应中心原子及与之直接相邻的化学键及原子,如此类推,可见随着直径d的增加,反应规则变得更加具体(图3)。该方法中反应规则的泛化水平是逆合成途径预测的关键技术之一,过于具体的规则会限制预测新路线的潜力,而过于笼统的规则可能会使预测偏离实际[37]。RetroRules[34]反应规则数据库按照SMARTS标准格式收录了超过40万条包含立体化学信息的反应规则,且每条反应规则均可以不同杂泛水平呈现。
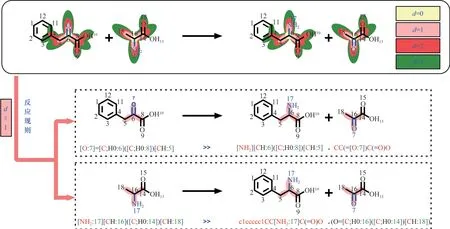
图3 反应规则的提取Fig.3 The extraction of reaction rules
1.4.2 专家反应规则集 自动提取的反应规则通常存在大量冗余,将降低途径延伸时的预测计算效率,且使产生的化合物和反应的数量呈现指数增长,造成组合爆炸问题。此时可通过人工精简产生规模较小但更精确的专家反应规则集,以限制途径延伸时反应的数量且减少网络规模,有利于提高途径延伸的计算效率及反应规则覆盖所有可能转换的全面性。RetroBioCat[37]人工构建了由83个反应组成的反应规则集,使用107个反应的SMARTS编码进行描述。Broadbelt[38]从基于原子映射的MetaCyc反应中自动提取反应规则,并通过人工精简获得最小但全面的1224条通用反应规则,经验证可唯一地覆盖所有常见的酶促转化,且能够重现KEGG和BRENDA数据库中超过85%的所有反应,有利于探索已知酶促转化的更大空间,加速生物合成途径的设计。
1.4.3 反应规则网络 代谢途径是生物体内的级联反应,基于代谢途径数据库可以将从数据库中学习到的所有反应规则按照反应规则网络(reaction rule network,RRN)的形式进行整合[16]。将各个反应规则均视为节点,若两个规则在已知途径上呈级联状态或具有形成级联反应的潜力,则在两个规则间添加边来连接,最终形成反应规则网络并将其应用在长途径化合物的预测上。例如,ReactionMiner[16]基于反应规则网络对衣康酸酯、柚皮素、1,3-丙二醇、木糖醇等高附加值化合物的生物逆合成途径进行了预测,发现可复原这些化合物的已知途径或预测出更短且生物学上更合理的逆合成途径。
1.5 酶序列的获取
化学催化和生物催化的主要区别在于生物催化采用酶作为催化剂,同样的,生物逆合成区别于化学逆合成的关键在于生物逆合成需要为预测的反应提供可能的催化该步反应的酶序列。然而,许多预测的新反应没有相关酶报道,预测与反应规则相关联、可催化新反应且与底盘宿主进化亲缘较近的酶的计算方法可大大加快生物合成途径的开发。
EC号是国际生物化学与分子生物学联盟(International Union of Biochemistry and Molecular Biology,IUBMB)中酶学委员会(Enzyme Commission)根据每种酶所催化的化学反应为分类基础制定的一套由四个级别组成的编号分类法,而反应规则同样体现了酶催化的功能,因此反应规则与EC号间存在关联,并通过查询EC号对应的酶序列可将反应规则与酶序列相关联,为预测出的新反应提供酶催化数据。
反应同源性是预测催化目标反应候选酶序列的基础,且不同酶序列预测工具在反应相似性矩阵的计算及反应的输入格式上有所区别。Yamanishi等[39]采用KEGG化合物ID作为输入内容,通过比较化合物间的转化模式,构建了E-zyme工具,可对化学反应分配EC号前三位数字。Goto等[40]对E-zyme工具进行了拓展并设计了E-zyme2工具,可基于RDM模式对底物-产物对进行全结构比对,当已知的相似反应与至少一个基因序列相链接时,可为输入的查询反应分配催化相似反应的酶序列。此外,Thornton等[41]开发了EC-BLAST工具,采用KEGG反应ID、SMIRKS反应规则和EC号作为输入内容,利用最大公共子图算法(maximal common subgraph,MCS)进行原子-原子比对(atom-atom mapping,AAM),并根据键的变化、反应中心及结构相似性从KEGG REACTION数据库中查询相似反应,以此为新反应分配前三位EC号,但EC-BLAST不能输出酶序列。Faulon等[42]开发了Selenzyme工具,采用SMIRKS化学模式语言作为输入内容,基于biochem4j图数据库,通过综合考虑序列相似性、酶序列的物理化学性质、酶来源物种与底盘宿主间的种群发生距离等方面来选择最优的候选酶序列。
BridgIT[43]考虑了辅因子在反应机制中的关键作用,是目前唯一可以区分不同酶催化反应机制且可预测从头设计反应的关联酶序列的方法。BridgIT采用SMILES表示的化学反应及BNICE.ch反应规则作为输入内容,可基于Daylight分子指纹计算目标反应与天然反应的Tanimoto相似性,并从KEGG中获取相似天然反应及其对应的酶EC号,进而提供一个候选酶EC号名单。对于KEGG(2011)中未注释酶序列且在KEGG(2018)中补充了注释的反应,BridgIT对其中90%的反应能够正确预测其相应的酶三级EC号。
基于以上工具,一些报道将酶数据库与反应数据库相结合,构建出反应规则-酶相互关联的综合数据库,方便生物逆合成算法的计算,如EnzyMine[34]将酶催化反应的特征与酶序列和结构注释相联系,构建了包含7767个EC号、267345条蛋白序列和9831个反应的综合酶-反应规则关联数据库。Fenner等[44]将从化学污染物生物转化反应数据库Eawag-BBD和KEGG数据库中获取的泛化生物转化规则与前三位EC号相关联,建立了具有316条反应规则-酶分类链接的enviLink数据库。
2 途径筛选指标与综合评价
逆合成网络中包含了大量预测途径,但并非所有预测途径都能够实现目标催化功能,目前已开发的工具所预测的途径假阳性过高,使得生物逆合成工具的应用仍不够普及,因此提高生物逆合成途径预测的准确率和可靠性是促进逆合成算法广泛应用的关键。从大量预测途径中推荐最佳候选途径,需要基于一些评价指标对预测途径进行评价、排序及筛选,目前已经报道的一些途径评价指标,主要从途径的理论可行性和与底盘宿主的适配性两方面对途径进行评价(图1,步骤8)。
2.1 途径的可行性
在途径筛选中,首先需要采用如底物相似性、热力学可行性、酶序列、途径长度等定量指标,对一些理论上不可行的途径进行排除。
2.1.1 底物相似性 考虑到生物逆合成算法采用酶-底物杂泛性假设,选择与已知底物结构相似性较高的输入化合物将更有可能被相应的已知酶催化;相反,若输入的查询化合物与化合物数据库中已知化合物的结构相似度较低,则相关联的已知酶对该查询化合物成功催化的可能性同样较低。基于化合物描述符,可以通过Tanimoto相似性等算法对预测的中间化合物与数据库中已知化合物间的结构相似性系数进行计算,从而在化合物数据库中检索结构相似化合物[7,12]。
2.1.2 热力学可行性 吉布斯自由能变化ΔG可表示反应的热力学势能变化,决定了酶促反应的方向性和效率,是检测和选择预测途径热力学可行性及评估生物合成途径热力学驱动力的重要手段。一些生物逆合成预测工具基于数据库中的反应吉布斯自由能数据或热力学计算工具检测途径的热力学可行性并对途径进行筛选,如Metabolic tinker基于CHEBi和RHEA数据库,使用此前报道的基于基团贡献(group contribution,GC)的热力学计算工具计算并评估途径的可行性[45];XTMS[9]基于MetaCyc database数据库提供的反应吉布斯自由能数据来评价途径可行性。此外,OptMDFpathway[46]利用基于约束的模型以途径的最大最小驱动力(max-min driving force)为优化目标,通过混合整数线性规划来识别具有最高热力学驱动力的途径,这类途径具有较高的代谢通量且对酶的表达强度要求较小。eQuilibrator3.0[47]可利用组分贡献(component contribution,CC)工具计算反应的生化平衡常数和ΔG,同时给出衡量预测不确定性的协方差矩阵用于基于约束的热力学模型计算。dGPredictor[48]基于KEGG数据库可利用基团贡献工具计算不同pH和离子强度下的代谢途径中酶催化反应的ΔG,且考虑了化合物结构中的立体化学信息,增加了热力学预测的精准度。
2.1.3 酶序列可行性 有无催化目标反应的酶序列对于预测途径的实现十分重要,尤其是对于基于反应规则预测的新反应。在延伸预测途径时可能一些反应规则不存在相关联的酶序列,此时预测出的新反应需要依靠人工查询文献以寻找酶序列,降低了设计的效率,因此在选择反应规则时应当增加与酶序列相关联的反应规则的权重,提高酶催化反应的可行性。
2.1.4 途径长度 途径长度是最直接的筛选指标,长途径意味着在底盘宿主中引入了更多的酶,从而使代谢负担增加,而结构复杂化合物需要经历多种后修饰过程,需要较长的生物合成途径才能完成修饰作用,因此需要对途径长度进行合理筛选。目前一些算法直接利用途径长度对预测途径进行打分和排序,而基于图论的生物逆合成预测工具通常采用混合整数线性规划等基于约束计算方法寻找底物到目标产物的前k条最短基元模式分析(elementary flux mode,EFM)[49]、最 短 碳 流 量 途 径(carbon flux path,CFP)[50]及最短活性途径(active pathway)[51]。例如,NICEpath[52]将反应物-产物对中保守的原子数量作为反应权重结合k-最短图搜索(k-shortest graph search)算法可实现KEGG中途径的筛选。PATHcre8[53]构建了包含可逆反应的双向图,采用Yen算法与PathLinker算法相结合的前K条无环最短路径算法筛选目标途径。
2.2 途径与底盘宿主的适配性
由于预测途径在底盘宿主中的实现会受到内源化合物及调控网络影响,从而呈现出偏离预测的现象,因此为了合理设计一个高效的异源生物合成细胞工厂,必须考虑外源反应在底盘宿主中特定内源性代谢网络影响下的稳定性,需要对预测途径与底盘宿主的适配性进行评价,以增加预测途径的可行性。OptStrain[54]、DESHARKY[55]、FMM[49]、Metabolic tinker[45]、GEM-Path[56]、XTMS[9]、MRE[57]、RetroPath2.0[11]等生物逆合成预测工具均对途径与大肠杆菌、酵母、蓝细菌等底盘宿主的适配性进行了探讨。
2.2.1 化合物毒性 中间化合物对细胞的毒性将妨碍途径中的酶在底盘宿主中正常表达,因此需要对化合物毒性进行预测,避免预测途径中包含高毒性中间化合物。通常采用化合物的半数抑制浓度(the half inhibitory concentration,IC50)作为化合物毒性的评价指标,表示一半细胞种群的生长受到抑制时的化合物浓度[58]。目前有一些收录了化合物毒性的 数 据 库,如TOXNET[59]、DSSTox[60]、T3DB[61]以 及RTECS[62]等,但其中的毒性数据多以动物细胞为对象,缺少对微生物细胞的毒性数据。目前已报道可预测化合物对微生物底盘宿主毒性的软件较少,其中EcoliTox[63]可基于化学结构与活性的定量关系预测中间化合物在大肠杆菌中的毒性,在多种逆合成工具中均有应用[9,11-12,64-65]。此外Toxicity Estimation Software Tool(TEST)工具也可实现对化合物毒性的预测。
2.2.2 代谢负担 代谢途径中引入的异源途径将增加细胞的代谢负担,从而妨碍细胞的生长和生产。DESHARKY[55]使用蒙特卡洛启发式算法对生物合成途径进行预测,同时对大肠杆菌中细胞资源和内源代谢情况进行建模,基于对核糖体和RNA聚合酶的消耗量计算异源代谢途径对底盘宿主的负担,从而可选取对宿主产生较小代谢负担的途径。
2.2.3 理论产量 通过构建包含底盘菌株内源化合物及预测途径相关化合物相对应的化学计量矩阵来构建GEM模型,并利用流量平衡分析(flux balance analysis,FBA)及基于约束的混合整数线性规划算法可计算途径在目标底盘宿主中化合物的理论产量,且常以产量最大化为优化目标来选择途径,这类生物逆合成预测工具包括OptStrain[54]、DESHARKY[55]、FMM[49]、GEM-Path[56]及XTMS[9]等。需要注意的是基于FBA的工具在评估途径时需要提供丰富的信息来为给定底盘细胞模型设定严格的反应通量边界,因而仅适用于大肠杆菌、酿酒酵母等经过充分研究的模式微生物。
2.2.4 内源起始化合物 底盘宿主内结构更简单的起始化合物通常具有更大的代谢通量,有利于增加目标产物的产量。SCScore[66]是一项衡量分子复杂性的指标,基于大量合成化学反应训练的神经网络对化合物分子的结构复杂性进行评分,从而有利于指导生物逆合成途径选择更简单的起始化合物。此外化合物官能团与反应中心碳原子之间的相对位置,将影响官能团的电子构型变化以及酶活性位点与底物的结合,进而影响化学反应的可行性。Lee等[67]基于ChemAxon Reactor工具通过比较所选氨基酸前体中官能团(氨基和羧基)与L-缬氨酸、L-亮氨酸和L-异亮氨酸的相对位置,为短链伯胺生物合成途径选择了最佳的氨基酸前体。
2.2.5 内源竞争途径 底盘宿主内源代谢网络可能会对异源生物合成途径产生竞争作用,进而影响目标化合物的生产。MRE[57]将预测出的异源生物合成途径整合到底盘宿主内源代谢网络中,并基于热力学可行性确定代谢网络中反应的方向,形成有向图,基于标准化的Boltzmann因子计算可对内源前体化合物(节点)进行转化的内源和异源竞争反应(边)的概率分布并由此进行赋权,以考虑特定底盘宿主中内源代谢反应对异源途径的竞争作用。
2.3 途径的综合评价
在实际情况中,常使用不同指标的加权组合来对途径进行综合评价,且一些基于综合评价获得的预测途径已被实验验证并用于微生物细胞工厂的构建。
XTMS[9]对食品工业中使用的昂贵风味成分树莓酮的生物合成途径进行了预测,并利用通量平衡分析(FBA)根据热力学可行性(吉布斯自由能)、酶性能(基因评分)、途径可行性(反应步骤的数量)、中间化合物的毒性及目标化合物产量等指标对预测途径进行了综合评价和排序,恢复了以香豆酰辅酶A为底物合成树莓酮的两步天然合成途径,同时考虑到作为底盘宿主的大肠杆菌中不存在香豆酰辅酶A,XTMS给出了从内源性化合物到树莓酮的生物合成途径。
PATHcre8可选择蓝藻为底盘宿主[53],预测从乙酰乙酰辅酶A到IPP、从甲羟戊酸到异戊二烯的生物合成途径及从可卡因到伪雌二醇辅酶A(pseudoecgonyl-CoA)的生物降解途径,并根据反应热力学可行性、途径中的潜在毒性产物(化合物毒性)、竞争反应消耗的途径中的中间产物(产物消耗)以及拷贝数等指标对途径进行综合评价,结果显示从乙酰乙酰辅酶A到IPP的天然途径在预测的前15条候选途径之中,经实验验证的基于磷酸异戊烯酯的(R)-甲羟戊酸到异戊二烯途径在候选途径中排名靠前,所预测的可卡因生物降解途径为尚未经过实验测试的潜在异源降解途径。
Ahsanul Islam等[68]利用ReactPRED和RetroPath2.0两种工具对苯、苯酚和1,2-丙二醇的生物途径进行预测,结合底物可行性和热力学可行性两个筛选指标,最终共获得49条生产苯、苯酚和1,2-丙二醇的预测途径,包含了从乙酸盐、葡萄糖和丙酮酸盐起始到苯、苯酚和1,2-丙二醇的106个反应,且25条预测途径完全由新反应组成,表明生物逆合成预测加速了潜在新反应的发现。
BioNavi-NP[7]对倍半萜类衍生物Sterhirsutin J和戊二酸的生物合成途径进行了预测,并结合底物相似性和途径长度两个指标对候选途径进行筛选,所得戊二酸的新生物合成途径已被实验验证[69]。Lee等[67]利用Park等[70]报道的工具对短链伯胺的生物合成途径进行了预测,结合底物相似性、反应位点相似性、热力学可行性、路径距离及酶与底盘的适配性五个评价指标对预测途径进行排序,所预测异丁胺生物合成途径在大肠杆菌中产量最高达到10.6 7g/L。Smolke等[71]利用BNICE.ch对那可丁衍生物的生物合成途径进行了预测,通过化合物引用次数、相关专利数对候选衍生物进行筛选,并结合热力学可行性、底物相似性、候选衍生物的生理功能对候选产物的预测途径进行评价,最终得到了(S)-四氢巴马汀、(S)-armepavine、(S)-laudanine和(S)-nandinine四种衍生物的候选途径,并在酿酒酵母中成功地进行了途径构建。
3 调控元件的设计和优化
微生物细胞工厂的设计不仅包括生物合成途径的设计,途径中所需的编码基因在底盘宿主中表达时还需要一系列必要的转录和翻译调节元件。这些元件将在一定程度上决定途径中酶的表达活性,并进一步影响菌株的生长和目标化合物的产量。因此需要对调节元件进行设计和优化,以精确控制酶活性且提供途径与底盘宿主的适配性,而人工智能的出现加速了元件从头设计的研究,并使人工定制遗传元件成为可能。由于真核生物转录和翻译调控十分复杂,对调控元件的研究集中在原核生物,尤其是大肠杆菌表达体系。
3.1 转录水平元件
启动子是在转录水平调控基因表达的关键元件,可驱动对基因表达的调控。先前寻找新启动子的研究主要集中在通过诱变或调控元件组合对已知启动子进行改造并形成启动子文库,结合人工智能手段对启动子的强度进行预测,以实现为细胞工厂提供不同转录强度的启动子元件(图4)。
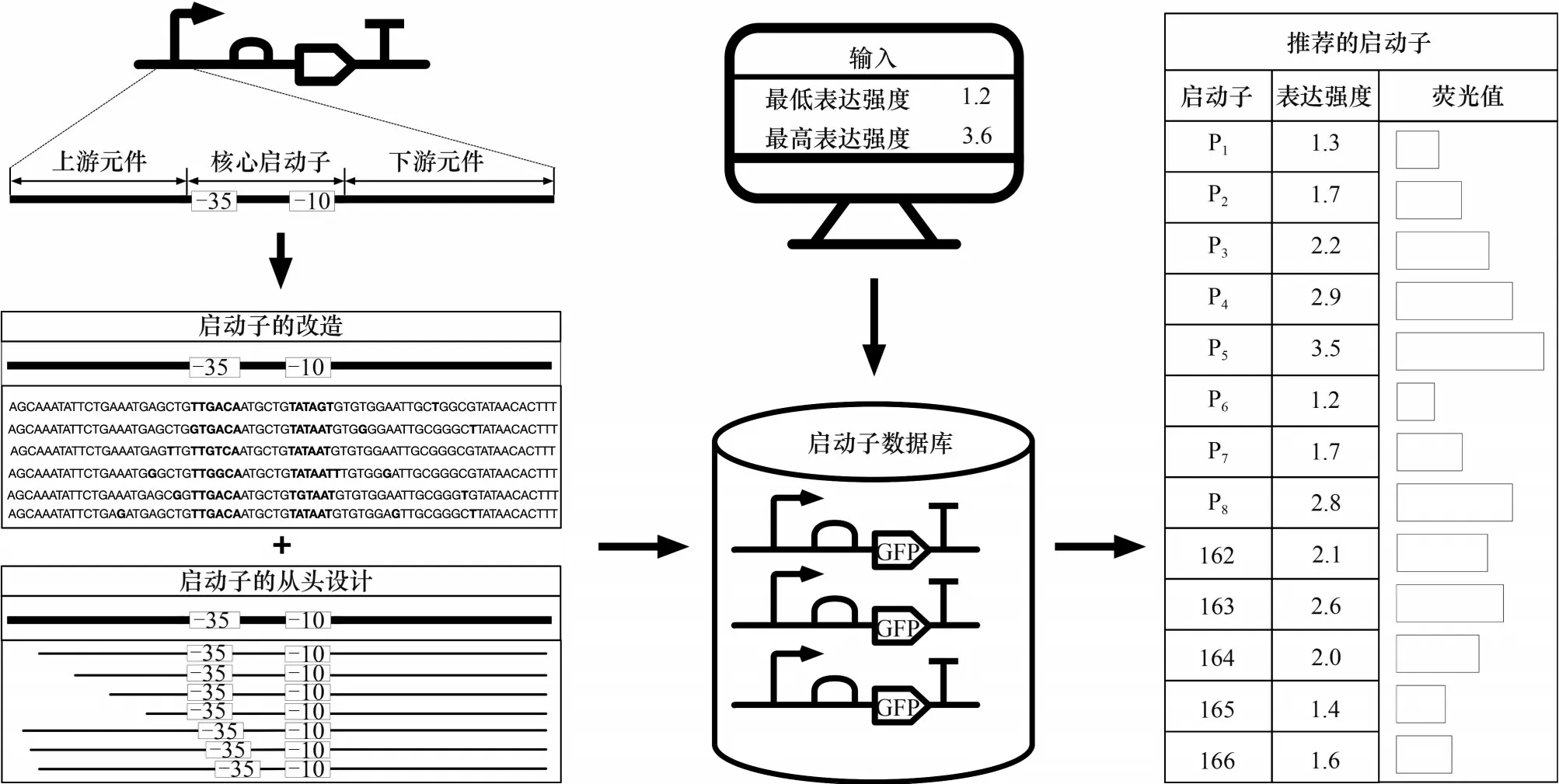
图4 启动子的设计Fig.4 The design of the promoter
在大肠杆菌的启动子设计研究中,SelProm[72]将拥有120个质粒的BglBrick文库中的诱导型启动子替换为组成型启动子,构建了10种不同表达强度水平的组成型表达质粒,覆盖的表达强度水平中最弱为未诱导的PlacUV5的1/5.6 ,最强比诱导的Ptrc高4.3 倍,最强与最弱表达强度水平之差为156倍,以良好的分辨率提供了广泛的表达水平。基于该数据,SelProm利用偏最小二乘回归(partial least squares regression)算法建立了预测选择模型,可对不同的质粒成分参数(启动子、抗性基因)下诱导型和组成型质粒的表达强度水平进行预测,并推荐目标表达强度水平相应的诱导型和组成型启动子,实验结果验证了启动子推荐工具的有效性。此外,Deng等[73]基于易错PCR技术对pTrc99a质粒上的Ptrc启动子进行诱变,产生了由3665个突变体组成的大肠杆菌人工启动子文库,所跨越的表达强度水平超过两个数量级,最强的启动子是1mmol/L IPTG诱导的PT7强度的1.52 倍。使用该合成启动子库作为输入数据集,构建并优化了基于XGBOOST机器学习算法的启动子强度预测模型,可对所设计的人工启动子的转录强度水平进行预测,且经比较发现,理性设计的一百个人工启动子的预测强度和实际强度十分接近(R2=0.88 ),从而验证了XgBoost模型在启动子表达强度的预测上的可靠性。
随着启动子突变体文库的增多,越来越多的启动子序列及其表达强度数据被公开报道,同时结合合成生物学及生物信息学领域的快速发展,启动子的从头设计成为可能。Wang等[74]基于生成对抗网络(generative adversarial networks,GANs)从大肠杆菌天然启动子中学习关键特征(k-mer频率、-10和-35基序及其间距限制),以捕获不同位置的核苷酸之间的相互作用,从而建立了大肠杆菌启动子的从头设计方法。人工启动子可基于大肠杆菌中的启动子活性和预测模型进行优化,两轮优化后高达70.8%人工启动子被实验验证了其调控转录水平的功能,且多数人工启动子与大肠杆菌基因组在序列上具有正交性。此外,其中一些人工启动子显示出与大多数天然启动子及其最强突变体相当甚至更高的活性,表明深度学习的方法可以为细胞工厂的设计提供更广泛的遗传元件来源。
真核生物的启动子设计研究报道较少,且集中于酿酒酵母表达体系,主要通过从酿酒酵母内源启动子中获取保守的模体(motif),并对模体之间的间隔序列进行设计,得到大型人工启动子文库,并结合人工智能算法建立启动子的预测模型。如Smolke等[75]以酵母内源TDH3启动子为研究对象,获取了TDH3启动子中转录因子结合位点、TATA框、转录起始位点等保守序列,对保守序列之间的间隔序列进行随机设计,得到超过675000条基于TDH3启动子的酵母人工启动子文库,测量了其中327000条序列的基因表达活性,并利用卷积神经网络算法建立了具有较高预测准确性的人工启动子表达强度预测模型。
3.2 翻译水平元件
对于原核生物而言,在翻译水平上的调控主要通过核糖体结合位点(ribosome binding sit,RBS)的设计来实现。RBS Calculator[76]、RBSDesigner[77]、RedLibs[78]、PartsGenie[79]等工具可预测RBS序列的翻译起始速率以估计给定mRNA序列的蛋白质表达水平,并被用于设计符合所需翻译起始速率的RBS序列。
例如,RBS Calculator[76]基于翻译启动阶段关键分子相互作用的吉布斯自由能建立了平衡统计热力学模型,通过将热力学模型与随机优化方法相结合,可设计具有特定的翻译起始速率或使得编码序列具有尽可能高的翻译起始速率的RBS序列,此外RBS Calculator还可以通过手动设计或复制强大的自然序列来设计比以前更强大的人工RBS序列。RedLibs[78]基于RBS Calculator可生成全局优化的简并RBS文库,以减少RBS文库中的冗余元件,且在文库规模尽量小的情况下包括更多中等或高强度的RBS序列,所得的兼并RedLibs文库中的核糖体结合位点样本能够以线性方式均匀覆盖整个翻译起始速率(rates for translation initiation,TIR)空间,充分满足不同强度RBS序列的选择需求。
此外,Ding等[80]将RBS的设计与生物传感器相结合,利用DNA微阵列构建了包含12000个RBS的葡萄糖酸生物传感器,通过荧光激活细胞分选(FACS)检测了生物传感器中绿色荧光蛋白的荧光强度,利用卷积神经网络(convolutional neural network,CNN)对其中7053个RBS的七个特征(RBS的GC频率,碱基A、T、C、G的频率,SDn的GC频率及SDm的GC频率)进行训练,建立了可预测RBS序列表达强度的神经网络模型CLM-RDR,能够快速确定与RBS序列对应的生物传感器的平均动态表达范围及其序列特征(图5)。
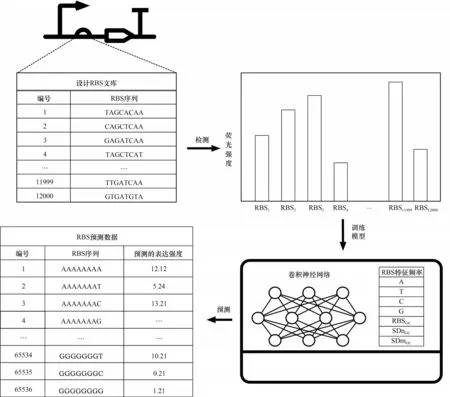
图5 核糖体结合位点的设计Fig.5 The design of the RBS
4 生物合成途径与调控元件的组合设计
在微生物细胞工厂的设计阶段需要确定所构建基因线路中启动子、终止子、标签、整合位点、载体等元件的组合方式,使目标途径能够发挥正常功能。在传统实验设计中由于不同特性的元件使得元件-途径的组合空间变得十分巨大,组合优化将使得实验量呈现指数级增加,在缺乏高通量构建平台的情况下难以实现,因此通常只能一次对某一类元件进行优化(one factor/one variable at a time,OFAT or OVAT),但元件间的内在联系和约束使所获取的组合方式只能达到局部最优。实验设计(design of experiments,DoE)是在生物工艺工程中广泛应用的高效探索大规模设计空间的系统方法,可以对组合空间进行优化、压缩,得到精简的组合空间,进而为获取全局组合优化提供了可能。在代谢工程及生物制造领域,DoE被用于优化实验条件以提高目标化合物产量(图6)。
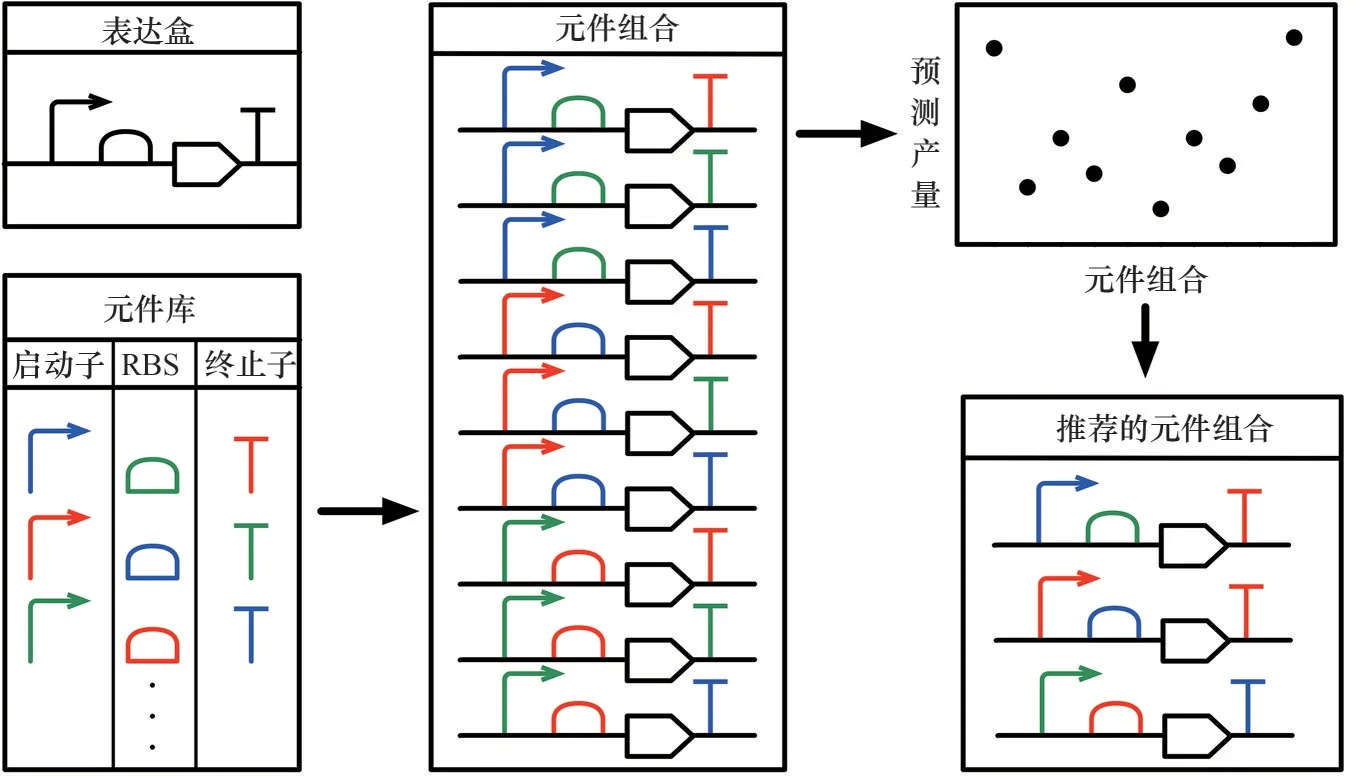
图6 实验设计Fig.6 Design of experiments
4.1 DoE的原理
DoE可以对有不同取值(levels)的因子(factors)的组合空间进行评价,并通过一些代表性实验来高效探索设计空间。DoE包括识别相关因子和对相关因子的取值组合优化两个过程。在微生物细胞工厂构建过程中,启动子、拷贝数、抗生素抗性等遗传相关变量以及碳源、氮源、添加物等培养基相关变量均可以作为具有不同取值的因子。而与预测途径及底盘宿主相关因子的识别和选择依赖于先验知识,其中在DBTL循环的早期阶段,先验知识较为匮乏,需要考虑较多的因子,并通过尽量少的DBTL的循环对设计空间进行精简。不同因子组合要实现的典型优化目标包括目标化合物产量的最大化(如增加得率、速率和产量)、使中间化合物毒性最小化等。
4.2 DoE优化途径与元件组合空间
Carbonell等[81-82]基于R程序包planor和DoE.base先后采用了以启动子强度、途径中基因位置次序及质粒拷贝数为因子和以底盘菌株和培养基组成为因子的方式对大肠杆菌中黄酮化合物(2S)-乔松素的产量进行优化,将(2S)-乔松素的产量提高了500倍。Singleton等[83]结 合DoE软件JMP Pro(SAS Institute Inc.USA)和人工神经网络(artificial neural networks,ANN),在摇瓶和96孔板两种生长环境条件下,将碳源、氮源、磷酸、维生素、氨基酸浓度等培养基相关变量作为因子,对热葡糖苷土芽孢杆菌(Geobacillus thermoglucosidans)的发酵培养基组分进行优化,实现热葡糖苷土芽孢杆菌利用己糖单糖和二糖进行生长,并可产生乳酸或乙酸盐。Radivojević等[84]结合机器学习和贝叶斯概率模型建立了自动推荐工具(Automated Recommendation Tool,ART),输入组学数据或启动子的组合可预测最终产量各种可能值的概率分布情况,同时基于逆向设计可提供使得目标化合物产量最高的候选组学数据或启动子组合,从而指导下一轮DBTL循环的实验设计,但ART目前仅支持单目标优化。Jensen等[85]将上述ART算法与酿酒酵母GEM、机器学习算法相结合,以启动子为因子对色氨酸(tryptophan)的生产进行优化,最终滴度和产量分别提高74%和43%。
5 结论与展望
生物制造作为制造业可持续发展的重要方向受到人们越来越多的关注,而微生物细胞工厂是生物制造的有力手段。当前持续积累的生物大数据极大地促进了计算机辅助设计工具的发展,对微生物细胞工厂的智能化设计将起到革命性的作用。本文依据细胞工厂在实际构建中的先后次序对细胞工厂中生物逆合成途径的预测与筛选、转录水平和翻译水平上遗传调控元件的设计、途径与元件的组合优化三个环节相关的智能设计工具进行了综述。
在生物合成途径的预测与筛选环节,生物逆合成算法基于泛化的反应规则扩展逆合成网络,并利用多种指标对预测途径的途径可行性和底盘适配性进行综合评价,给出最具实际可行性的推荐途径,帮助人们进行已知途径化合物的途径优化及提产和未知途径化合物的途径预测及设计。此外,蒙特卡洛树等人工智能算法的引入为生物逆合成算法的发展提供了新的思路。但值得注意的是,当前生物逆合成工具预测途径的假阳性率仍然较高,其主要原因在于途径评价算法不能充分模拟底盘宿主对途径的选择,如不能精确计算化合物对指定微生物宿主的毒性、未考虑化合物及酶的区室化对途径表达的影响以及缺乏酶催化底物杂泛性数据等。目前AlphaFold2[86]和RoseTTAFold[87]的出现可实现基于序列预测蛋白质晶体结构,为酶与底物的适配性问题提供了解决思路。
在遗传调控元件设计环节,主要研究对象是原核生物的启动子和核糖体结合位点设计。对原核启动子的设计主要从对已有启动子进行改造(诱变、易错PCR等)或从头设计两种手段获取新启动子,并基于启动子调控的荧光蛋白的表达强度数据建立预测模型及人工启动子库,为所需表达强度提供推荐的启动子序列。由于大肠杆菌等原核生物中启动子的长度较短,一般小于150bp,而酵母等真核生物的启动子较长,通常为数百个核苷酸,且调控机制更为复杂,因此开发适用于真核生物启动子设计和表达强度精准预测工具是遗传调控元件设计的重要挑战。此外,原核生物核糖体结合位点的设计主要基于热力学模型,可计算RBS序列的翻译起始效率,并提供推荐的RBS序列。值得注意的是,对这两种遗传调控元件进行设计的工具均很少考虑目标序列的实际表达环境参数(菌株、pH、质粒类型、标签等),减少了推荐的遗传元件可靠性。
在途径与元件组合优化环节,DoE方法简化了遗传元件与途径的组合空间,可以在多轮迭代设计中优化途径表达及目标产物的产量,但DoE方法难以充分考虑途径设计及构建中的相关因子,如底盘宿主中可能影响途径表达的干扰因子,使得DoE推荐的组合方案具有一定的局限性。
此外,目前微生物细胞工厂相关的设计工具只能相对独立地进行特定环节的设计,不能实现微生物细胞工厂的一站式自动化设计“流水线”,需要靠人力来完成各部分设计工具间的连接,因而不能有效地提高细胞工厂的设计效率,通过整合现有工具资源或创制新的工具、统一接口、建立标准化的自动化设计工作站,将是微生物细胞工厂智能设计的重要发展方向。
猜你喜欢
小猕猴智力画刊(2022年3期)2022-03-29
中学生数理化·中考版(2021年12期)2021-12-31
中学生数理化·中考版(2021年11期)2021-12-06
数学小灵通(1-2年级)(2021年4期)2021-06-09
中学生数理化·七年级数学人教版(2019年10期)2019-11-25
中学生数理化·高一版(2018年9期)2018-10-09
湖南教育·C版(2018年3期)2018-06-05
Coco薇(2017年11期)2018-01-03
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
