
咪唑类离子液体催化四氢噻吩氧化脱硫机理*
2022-01-10 07:56:46张建亮杨德伟王新伟林日亿
油田化学 2021年4期
陈 凯,张建亮,杨德伟,王新伟,林日亿
(1.中国石油大学(华东)新能源学院,山东青岛 266580;2.南方海洋科学与工程广东省实验室(湛江),广东湛江 524002)
0 前言
随着常规原油资源减少,稠油成为各国寄予厚望的主要能源之一[1]。但是稠油中存在较多的有机硫化物,稠油脱硫成为人们关注的热点。加氢脱硫(HDS)是目前工业上应用最广泛的脱硫方式[2],在稠油水热裂解过程中通过加入供氢剂生成H2S,从而去除重油中的硫[3]。但是噻吩类化合物的空间位阻大,加氢活性很低,加氢脱硫方式较难脱去噻吩类化合物中的硫[4]。此外,HDS 存在难以避免的成本大、能耗极高、腐蚀设备等问题。
离子液体是一种绿色溶剂,同时具有萃取剂和催化剂的作用。在离子液体的催化作用下,硫化物被氧化为砜或亚砜,再被离子液体作为萃取剂分离出来[5]。离子液体在石油脱硫中有着广泛的应用[6]。Lo 等[7]利用[C4mim]PF6和[C4mim]BF4对油品脱硫,首次将萃取与氧化过程结合起来,发现相比于萃取脱硫的方式,加入氧化剂进行氧化萃取脱硫的效率要高一个数量级。Mochizuki[8]研究了不同烷基链长的离子液体对模型油的氧化脱硫效果,发现随阴、阳离子的烷基链的增长,特别是阳离子的烷基链增长,离子液体的脱硫效率明显增大。
离子液体不同的阴、阳离子影响脱硫的效率。胡松青等[9]研究发现,阳离子相同的离子液体,改变阴离子种类,脱硫效率有显著的不同,其中[AlCl4]-离子液体>[PF6]-离子液体>[BF4]-离子液体。针对不同的噻吩类化合物,同种离子液体的脱硫效率也不同。Esser 等[10]发现离子液体对二苯并噻吩(DBT)的萃取效果最好,对于常见的噻吩类化合物,萃取效率依次是DBT>苯并噻吩(BT)>4,6-二甲基二苯并噻吩(4,6-DMDBT)>噻吩(TS)。离子液体用于稠油改质降黏时,主要是发挥萃取或者辅助氧化的作用。范洪富等[12]合成咪唑氯化铁盐类离子液体,用于实验室稠油的改质降黏,发现离子液体质量分数在5%时,能将稠油黏度降低80%以上。咪唑氯铝酸型(Lewis酸)离子液体是最早用于脱硫的离子液体,其脱硫效果较好,高温下较为稳定,但是容易与水发生反应;其他阴离子的咪唑类离子液体,如BF4-、PF6-遇水则相对较为稳定,但它们易与非含硫的芳香化合物结合,导致油品辛烷值降低[13]。更优脱硫效果的离子液体结构有待研究。在微观层面,Cheng等[14]根据DFT理论中的GGA-PW91方法,模拟了燃料油加氢脱硫过程中离子液体的催化作用;吕仁庆等[15]利用分子模拟发现了咪唑类离子液体与噻吩之间主要是阳离子与噻吩之间的C—H···π作用。Zhang 等[16]研究发现离子液体能使DBT的极性增加,从而使催化效率得到提高。
许多专家学者已经对离子液体脱硫进行了大量的研究。然而目前的研究较少从微观层面解释离子液体催化脱硫机理;大多数研究以稠油为研究对象,但由于稠油成分复杂,在对反应机理的研究中存在不确定性。四氢噻吩(THT)性质与其他噻吩类化合物相差较大,但是针对THT 的研究也较少。本文以THT 为模型化合物,以H2O2为氧化剂,探究了THT 在咪唑氯铝酸盐离子液体催化下的氧化脱硫过程;探究了离子液体及其阴、阳离子与THT 形成的稳定结构,验证了不同侧链长度对该结构稳定性的差别,并作出分子层面的解释;根据马林肯电荷布局、前线轨道分布情况,解释了离子液体对THT氧化过程的催化作用。
1 计算模型及计算方法
本文的模型构建、几何优化以及模拟计算使用戴尔T7910工作站,八核十六线程,搭配Windows 7 Professional 系统。计算软件采用Materials Studio 8.0。THT和离子液体以及其他反应物、产物的结构优化使用密度泛函理论(DFT)中的广义梯度近似方法(GGA),选用BLYP密度泛函处理交互相关能,并对每一步的优化结构进行了振动频率的验证和分析,确定结构优化的稳定性和科学性。
计算中采用双数值型基组加轨道极化函数(DNP)处理价电子波函数。计算中原子核处理(Core Treatment)选择All Electron Relativistic 处理体系中所有的电子,所有的计算都采用了电子自旋极化。具体步骤如下:首先构建各粒子的结构,包括THT、离子液体及其阴离子、阳离子,用Dmol3模块分别进行结构优化,综合考虑计算效率及计算精度,本文所有体系的精度设置为Medium,轨道热占据采用默认的0.005 Hartree,能量标准采用2×10-5Hartree,梯度采用4×10-3Hartree/Å、最大位移使用5×10-3Å,SCF迭代精度采用1×10-5Hartree。
根据文献[15]探究各种相对位置下构成的相互作用结构,采用能量最低的原则,选取每种相互作用下的最稳定的结构,作为后续的模拟计算。为了分析粒子之间的相互作用,定义结合过程中的结合能如下:
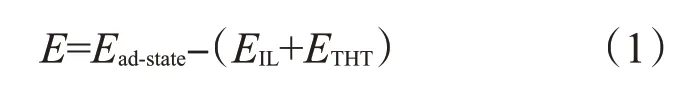
其中,Ead-state、EIL、ETHT分别表示结合后结构、离子液体、THT 的能量,E表示结合过程的结合能,其绝对值越大,表示对应的结构越稳定。
为了在分子水平解释离子液体的催化机理,还进行了马林肯电荷的计算以及HOMO 和LUMO 轨道能量的计算。计算时采用DMol3 模块中Orbitals的功能,在properties 项中勾选HOMO、LUMO 选项。其他计算参数设置与结构优化中的设置参数保持相同。
最后,通过线性同步度越方法(LST)和四级同步度越方法(QST)搜索过渡态,对整个氧化反应过程的原子转移情况以及能量变化情况进行探究。模拟计算时,利用优化后的结构,先用Reaction Preview 定义原子配对并预览反应过程。然后选择TS search,其他计算参数设置与结构优化中的设置参数保持相同。完成后利用TS Optimization工具对过渡态进行精修,保证反应过程只有一个虚频,同时使得过渡态能量更加精确。
2 结果及讨论
2.1 THT-ILS的结构优化
在实际工况中,离子液体的用量相对反应物较少,故它在反应过程中主要以离子形式存在。以下分别考虑了离子液体的阳离子、阴离子、离子液体整体的不同催化情况。
首先构建了离子液体、阳离子[EMIM]+、阴离子[AlCl4]-、反应物H2O2和THT、产物H2O 和THTO(亚砜)的模型结构,并进行结构优化;将离子液体与反应物、产物以不同相对位置作用,试验不同结合形式,进行结构优化,根据能量最低的原则,找到他们最稳定的相互作用结构。图1分别包括了氧化前的THT与离子液体的阳离子、阴离子以及离子液体整体的稳定作用结构,图2 分别包括了氧化后的THTO 与离子液体的阴、阳离子以及离子液体整体的稳定作用结构。
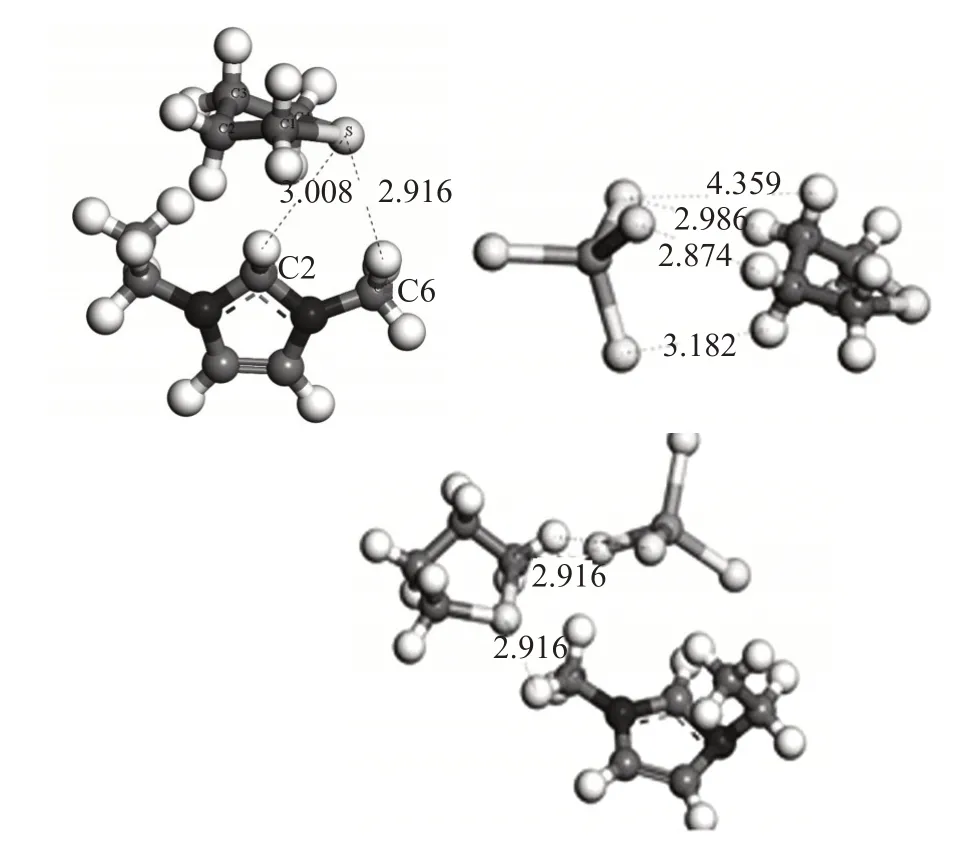
图1 THT与离子液体的阳离子、阴离子以及离子液体整体的作用结构
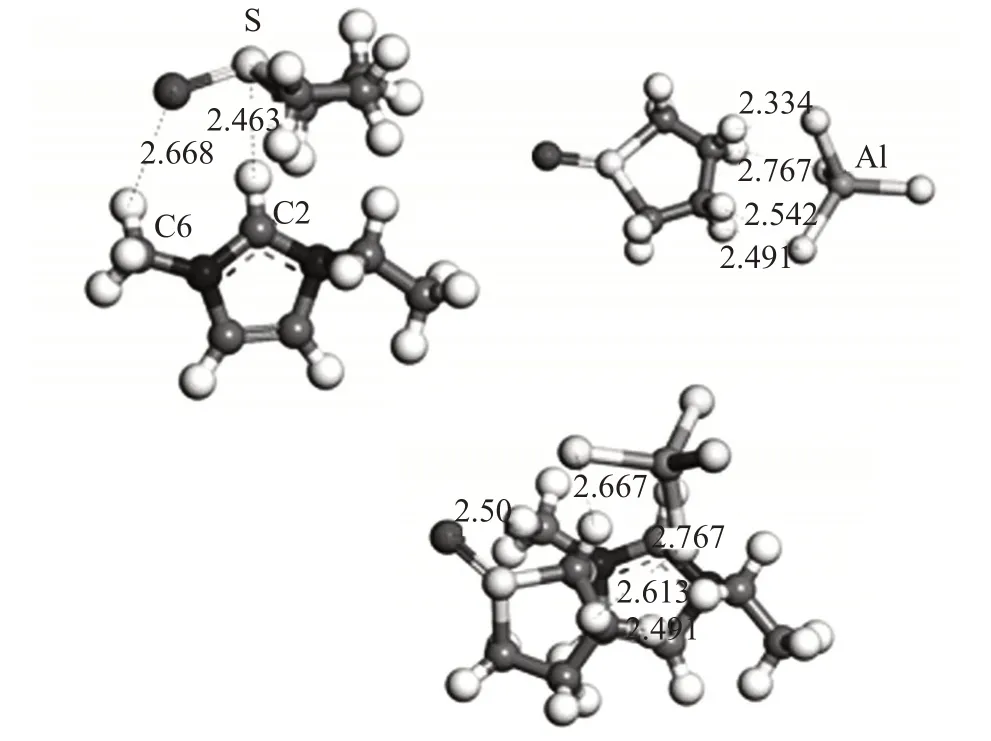
图2 THTO与离子液体的阳离子、阴离子以及离子液体整体的作用结构
在THT 与咪唑阳离子的稳定结构中,THT 的S原子和[EMIM]+的2位C、6位C上的H原子存在氢键,该C 原子上形成的C—H 键是δ单键,与THT 上的S 的孤对电子相互作用。根据文献,咪唑阳离子与噻吩类化合物之间的作用力还包括S原子与咪唑环之间的π-π作用[17]。
THT 上的C2 和C3 上的H 原子,与离子液体的阴离子[AlCl4]-中的Cl 原子,在稳定结构中最近距离为2.874 Å和2.986 Å,且键角接近180°,形成了分子间氢键。Al原子的3s轨道被激发,形成sp3杂化,四个Cl原子地位相同,结构完全对称。所以形成的Cl—H氢键比较接近,但是键角略有差异,主要是因为THT与[AlCl4]-所形成的稳定结构并非是完全对称的。而作为整体,离子液体与THT的相互作用主要表现在THT 上C4 上的H 原子和与[AlCl4]-上的Cl 原子之间的氢键作用,以及THT 上S 原子与[EMIM]+上C6位上的H原子之间的氢键作用[17-18]。
THT 在被离子液体氧化成亚砜(THTO)之后,它与离子液体的[AlCl4]-之间的作用方式与被氧化前的THT 的作用方式基本相同,但氢键的长度变短;这说明THTO与阴离子的氢键作用增强了,这与结合能对比的结论相同;THTO 与离子液体阳离子之间的作用,与氧化前S原子上形成的氢键相比,氧化之后作用形式发生了变化,主要增加了O原子上形成的氢键,氢键长度变短2.463 Å,由于O 原子的电负性与Cl 原子相比更大,所以引入O 原子后,形成的氢键更强。THTO 与离子液体作用时,其O 原子和侧链甲基之间形成氢键,两者之间的主要作用以THT和阳离子之间的作用为主,它们之间除了氢键作用外,还存在咪唑环上大π键与δ单键的作用[17]。改变咪唑环侧链的长度,即替换[EMIM][AlCl4]咪唑环的3-乙基,将侧链的C原子数依次改变为2、3、4、5、6,分别对应离子液体[EMIM][Al-Cl4]、[PMIM][AlCl4]、[BMIM][AlCl4]、[AMIM][AlCl4]、[HMIM][AlCl4]。用以上相同的方法,分别找到每种离子液体与THT和THTO的稳定结构,并计算其结合能,结果如图3所示。
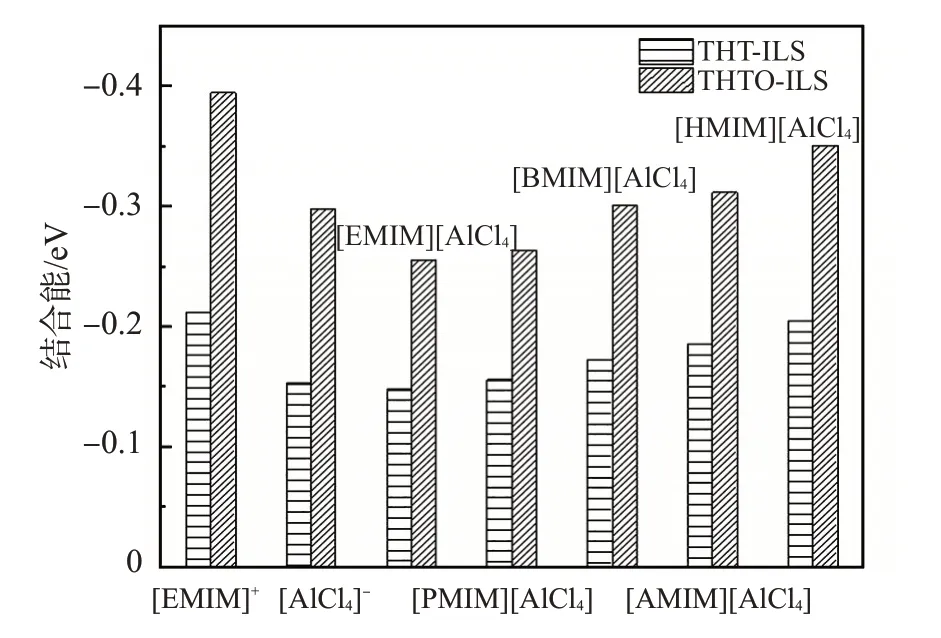
图3 不同离子液体与THT、THTO之间的稳定结合能
以[EMIM][AlCl4]为例,比较离子液体中[EMIM]+、[AlCl4]-、[EMIM][AlCl4]与THT 的结合能发现,THT 与阳离子相互作用时,放出的能量最多;而离子液体作为整体结合时,结合能最小;随着侧链长度的增加,离子液体与THT相互作用放出的能量越来越大。这是因为,当侧链的长度增大时,侧链的电子流向咪唑环,咪唑环上自身的负电荷量增加,使得THT上的S原子出现更明显的电荷富集现象,S原子的电荷密度变大,在氧化的过程中更容易给出电子,即更容易被氧化。同时,阳离子的体积增大,则离子液体整体内部的库伦作用就会减弱,阴阳离子之间的堆垛结构就相对较松,使得THT 能够更容易插入其中。这也可以推出离子液体作为氧化产物萃取剂的原因。在萃取过程中,离子对氧化后的氧化产物具有相对更大的结合能,更容易将氧化产物萃取到离子液体层。同时,在反应系统中将反应产物萃取出来之后,也进一步促进了反应平衡向正反应方向移动。这与文献[18]中的变化规律一致。
2.2 马林肯电荷分析
根据THT和离子液体的稳定结合结构,以下从其稳定结构的马林肯(Mulliken)电荷布局的变化情况,探究THT在与离子液体相互作用之后电子的转移情况,从分子角度解释离子液体在THT被氧化的过程中所起的催化作用,以THT与[EMIM][AlCl4]的相互作用结构为例。表1列出了THT在与离子液体形成稳定结构后的Mulliken电荷分布。从表1可以看出,单独游离状态下的THT 分子,其S 带有0.288 e的负电荷,靠近S原子的C1和C4均带0.116 e的负电荷,C2和C3均带0.162 e的负电荷,这是由于硫原子本身的吸电子效应,使得Cl、C4 所带的负电荷较C2、C3 的更少;而环上的H 原子均带有正电荷,自由状态下的THT 分子电荷整体为0。与自由态的THT 分子相比,当THT 与离子液体作用时,S和H均失去电子,C均得到电子,THT整体电荷量减少0.256 e,说明在整个作用过程中电子通过THT分子转移到离子液体。

表1 稳定结构下四氢噻吩分子的Mulliken电荷分布
2.3 前线轨道理论分析
为了探究离子液体对THT 催化作用的微观机理,根据分子的前线轨道理论,探究稳定结构中的最高占据轨道(HUMO)和最低未占据轨道(LUMO),对其中的电子转移趋势进行了分析。图4 为离子液体以及THT 的前线轨道分布。通过模拟计算5种不同链长的咪唑氯铝酸离子液体以及THT的最高占据轨道(HOMO)和最低未占据轨道(LUMO)的能量特征值,结果如表2 所示。根据图4 和表2 数据,THT 的最低未占据轨道(LUMO)和各离子液体的最高占据轨道(HOMO)之间的能隙,最大为3.82 eV,最小3.71 eV,而5 种离子液体的最低未占据轨道(LUMO)和THT 的最高占据轨道(HOMO)之间的能隙最大为2.29 eV,最小为2.16 eV,因此,离子液体的最低未占据轨道(LUMO)较容易从THT 的最高占据轨道(HOMO)得到电子。这与文献[19]中的研究一致,同时也与2.2 节的结论相吻合。总体上看,随着侧链长度的增长,离子液体的最高占据轨道(HOMO)能量逐渐升高,而最低未占据轨道能量(LUMO)轨道逐渐降低,这是由于侧链越长,侧链向咪唑环提供电子的能力逐渐加强。而在催化过程中,S原子发生电荷富集,咪唑环上电荷量越多,S 原子电荷富集现象越明显,共价性减小,S—C键被减弱,那么S原子在被氧化过程中就越容易给出电荷。即离子液体通过亲核过程活化了THT分子,使其还原性得到增强,更容易被氧化。说明离子液体对H2O2氧化THT起到了催化作用。
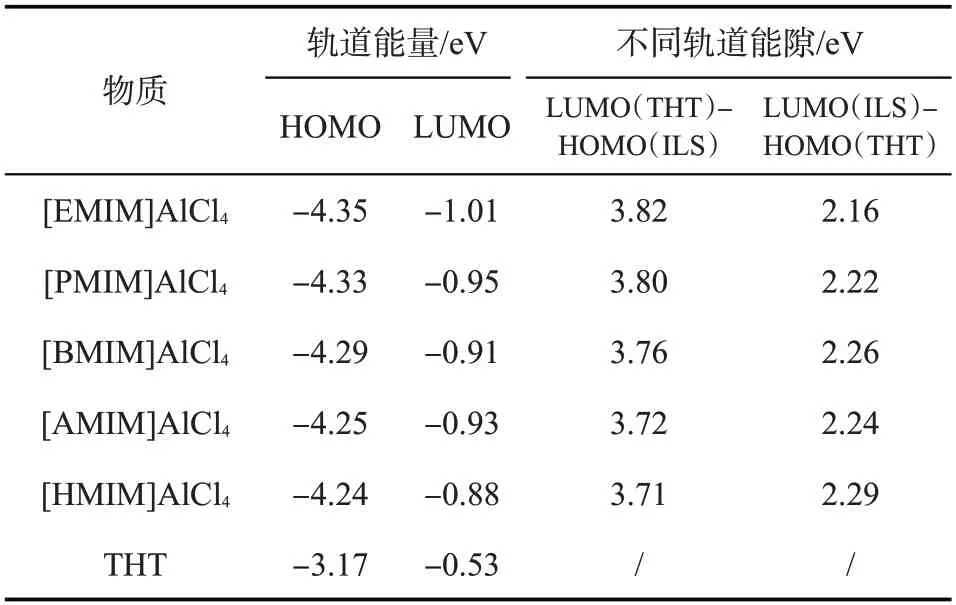
表2 各离子液体以及THT的HOMO和LUMO特征值
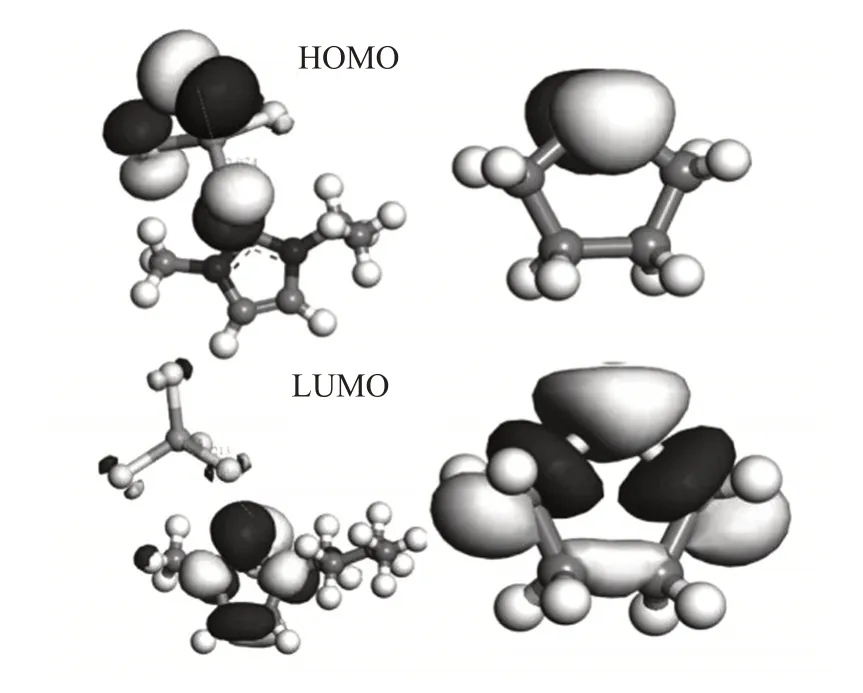
图4 离子液体以及THT的前线轨道分布
2.4 四氢噻吩氧化过程
H2O2对THT 的氧化过程:THT+H2O2→RS2→TS→THTO+H2O。H2O2氧化THT的反应过程如图5所示。反应初始,THT中的S和H2O2中的O相距较远,为3.86 Å,化学键的长度通常都在3 Å以下,所以此距离下通常无法形成化学键。H2O2的O—O键键长为1.48 Å。反应开始进行时,THT 和H2O2逐渐靠近,S 原子和H2O2中的一个O 原子开始有了电荷的转移,具体表现为S 原子上的孤电子对向这个O 原子转移,H2O2发生翻转,且变得更加不稳定;O—O键变长,而其中一个O 原子与S 原子的距离更近。如RS2所示,此时O—O键长为1.70 Å,而S、O原子之间的距离为3.09 Å,同时伴随电荷的转移,S、O原子之间已有成键条件。从RS2 到过渡态TS 的过程中,O—O键已趋于断裂,S、O原子相距2.59 Å,此时的系统有了唯一的虚频,它来自于H2O2中O—O 键的伸缩振动,频率为-467.4 cm-1,O—O 键不断伸缩振动,最终发生断裂,并且由S 原子的孤电子对转移,形成S=O 双键,这个O 原子上的H 原子靠近H2O2的另一个O 原子,并最终生成一个水分子。THTO与H2O的距离变大,并趋于稳定,THT的氧化过程结束。
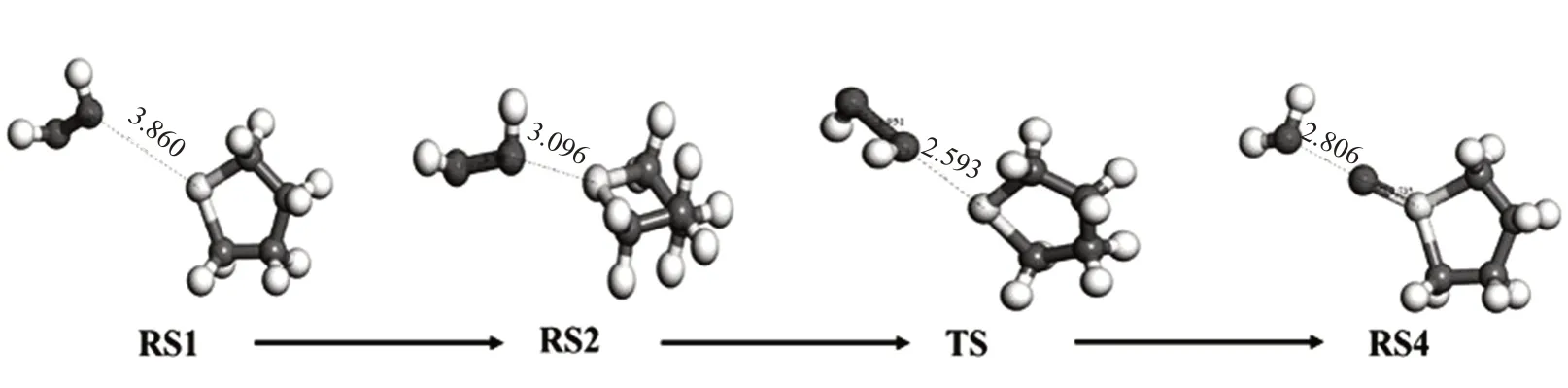
图5 THT被H2O2为THTO的反应过程
在THT氧化过程中,通过对比是否加入离子液体时的氧化情况,分析离子液体在氧化反应中所起到的催化作用。以下仍然以[EMIM][AlCl4]为例进行说明。
反应过程如图6 所示,THT 和H2O2在反应过程中的状态变化与不加离子液体时相似,但是每个状态对应的能量却相差较大。把每个状态的能量通过分子模拟计算出来,不加离子液体、加入离子液体[EMIM][AlCl4]两种情况下的反应能量变化如图7所示。
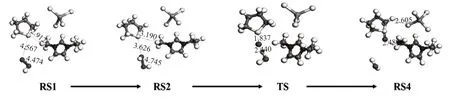
图6 [EMIM][AlCl4]催化下THT被氧化为THTO的反应过程

图7 [EMIM][AlCl4]催化下四氢噻吩被氧化为THTO的能量变化
由图7 可以看出,离子液体的存在能显著降低反应过程的能垒,在形成中间体RS2 时,加入离子液体使得该反应的吸热过程变为放热过程,反应更容易发生。在反应的第三步,也就是H2O2的O—O键断裂,S原子发生孤对电子转移时的状态,此时为反应的过渡态,同样地,系统出现了唯一的虚频。加入离子液体后,过渡态的能量从不加离子液体的292.25 kJ/mol 降到130.88 kJ/mol,反应能垒也降低了约55%,反应变得更容易进行。
咪唑类的离子液体种类众多,改变离子液体咪唑环的侧链长度,就能得到不同种类的离子液体。将不同种类的离子液体分别加入THT 与H2O2的氧化体系中,THT 被氧化的反应过程类似,但是在反应过程中各个阶段的能量及能量变化不同,氧化过程中各阶段的能量变化如表3所示。

表3 不同阳离子的离子液体催化下驻点结构能量
可以看出,改变离子液体的阳离子、咪唑环侧链C 原子数分别为2、3、4、5、6 个,即随着侧链取代基分别为乙、丙、丁、戊、己基时,反应的能垒有下降趋势,说明侧链越长,对反应的促进效果越明显,这和前文前线轨道的计算分析结论吻合,并与文献[8,20]结论一致。在反应过程中,离子液体与H2O2作用后,H2O2的O—O 键变长,H2O2明显被活化,氧化性也得到增强。离子液体对反应的催化作用不仅表现在对THT上S原子电荷的影响,还对反应物H2O2有活化的作用,在反应过程中使O—O 键的键长变长,从而使H2O2更加活跃。
3 结论
在离子液体与THT的各种稳定结构中,THT与阳离子之间的氢键作用主要表现在S 原子与咪唑的2、6号位C上的氢键作用。THT被氧化成THTO后,O 原子更强的电负性使THTO 与离子液体的作用增强,阳离子的侧链越长,相互作用的结构越稳定。
针对不同阳离子的离子液体,阳离子的侧链越长,由于侧链供电子能力的增强以及离子液体整体内部的库伦作用的减弱,离子液体对THT 以及THTO的作用力增强。
THT与离子液体相互作用时,整体上电子通过THT转移到离子液体,但是THT上的S原子与相邻的C原子会发生电荷富集。THT上S原子电荷量增加,S 原子与C 原子之间的电荷偏移减少,S 原子与C原子之间的相互作用减弱,THT更容易被氧化。
离子液体的加入降低了THT 氧化过程的能垒55%以上。离子液体不仅对THT电荷结构有影响,还对H2O2有活化作用,且阳离子侧链越长,效果越显著。
猜你喜欢
中国民间疗法(2021年17期)2021-11-04 08:39:44
山东农业大学学报(自然科学版)(2021年3期)2021-07-29 03:07:02
世界农药(2019年2期)2019-07-13 05:55:12
粘接(2017年4期)2017-04-25 08:37:20
兽医导刊(2016年6期)2016-05-17 03:50:16
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年10期)2016-01-17 08:56:47
中国卫生标准管理(2015年2期)2016-01-14 03:41:34
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
西南军医(2015年2期)2015-01-22 09:09:22
