
团簇Mo3S4析氢性能的密度泛函理论分析
2022-01-10 09:11朱依文方志刚曾鑫渔毛智龙郑新喜
西安工程大学学报 2021年6期
朱依文,方志刚,王 倩,曾鑫渔,毛智龙,郑新喜
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
0 引 言
过度使用化石燃料导致的能源危机和环境污染问题促使研究人员迫切需要寻找高效、清洁和可持续的能源。氢能以其绿色环保、可持续发展等优势在众多新能源中脱颖而出[1]。尽管氢能发展前景广阔,但我国整体技术水平仍显不足,因此高效析氢催化剂的制备备受关注。铂金属资源珍稀而未能普遍应用,非铂基材料应用中能量损失相对较大,因此寻找低成本、活性位点更多的高效电催化析氢催化剂成为近年来的研究热点[2-4]。
钼基硫化物不仅具有很好的稳定性和催化活性,而且具有超高的磁导率和电阻率[5-8]。GUILLAMN等研究了异硫化H2活化机制下二方体团簇Mo3S4对氮苯的催化加氢作用,通过2个相互连接的催化循环生成二苯肼和苯胺[9],其中制备团簇Mo3S4采用的硫基活化氢气方法亦可应用于其他加氢反应。SEO等研究发现团簇Mo3S4作为仿生电催化剂,在与NaTaO3对水进行光催化分解成H2和O2的共催化反应中,极大地提高了NaTaO3的光催化活性[10]。DUVAL等研究发现团簇Mo3S4与Mo18环组合后能够增强其诱导电催化行为[11]。WANG等将单层2H-MoS2结构域叠加上多层1T@2H-MoS2制备了悬空键和晶格失配更少的MoS2光电探测器,为今后开发更强光响应的光电探测器奠定了基础[12]。目前大多数研究均为宏观实验,理论研究相对较少。因此从理论和实验方面进一步探讨团簇Mo3S4的性质十分必要,本文将对团簇Mo3S4模型的催化析氢活性进行分析。
1 实 验
1.1 模型设计和计算方法
参考文献[13],使用高压合成技术制备新多晶型Mo3S4,按照Mo∶S=3∶4的基础化学计量比,依据拓扑学原理[14],设计局域模型团簇Mo3S4。以五棱双锥型、戴帽四棱双锥型和戴帽三角双锥型为初始构型,通过改变不同原子间的位置关系设计其构型方案,使用Guassian09程序计算单、三重态下的初始构型,运用密度泛函理论[15-17],在B3LYP/Lan12dz水平下进行优化,排除虚频和相同构型后得到8种稳定存在的优化构型,对各构型中Mo、S原子采用文献[18]中含相对论校正的有效核电势价电子从头计算18-eECP双ξ基组(3s,3p,3d/2s,2p,2d),并对S原子加极化函数ξS,d=0.55[19]。对水分子的O、H原子则均采用Dunning/Huzinaga双ξ基组(9 s,5p/3 s,2 p)。
1.2 析氢反应机理
以团簇Mo3S4(在反应式中以M表示)作为模拟对象,其催化机理如下。
第一步 团簇吸附氢原子:

(1)
第二步 析出氢气(该反应存在2种反应途径):
1) 电化学解吸
M—Hads+e+H2OM+H2+OH-
(2)
2) 化学重组:
(3)
2 结果与讨论
2.1 团簇Mo3S4的优化构型
采用上述方法对团簇Mo3S4进行优化计算、排除虚频和相同构型后,确保每个优化构型真实存在且相互不重复,最终得到单、三重态共8种优化构型,如图1所示。由于立体形态构型存在空间结构差异,将团簇Mo3S4的优化构型分为3类:五棱双锥型(1(1)、2(1))、戴帽四棱双锥型(2(3)、3(1)、3(3)、4(3))和戴帽三角双锥型(1(3)、4(1))。右上角的数字表示构型所属重态。为方便比较,将能量最低的构型1(1)作为参考构型,将其能量值设为0 kJ/mol,其他单、三重态各构型能量以此为标准重新计算,能量由低到高依次为:1(1)<1(3)<2(1)<2(3)<3(3)<4(3)<3(1)<4(1)。
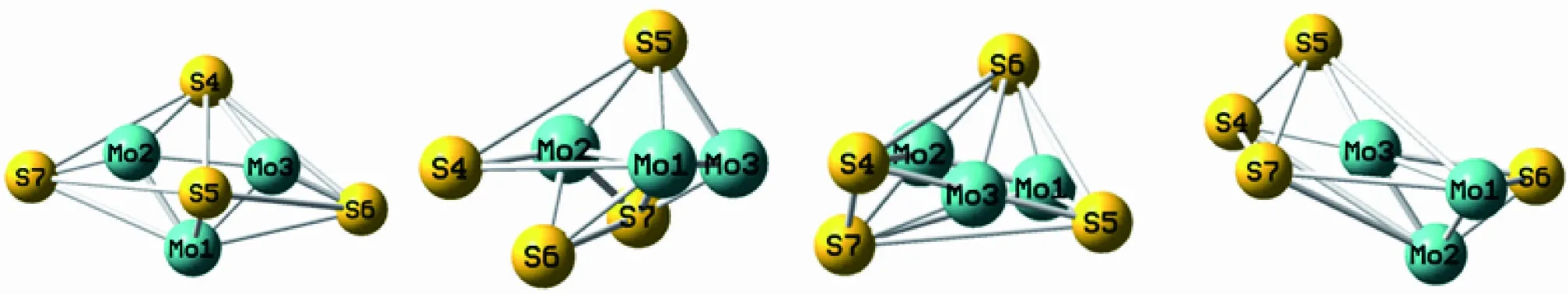
1(1)(0 kJ/mol) 1(3)(10.054 kJ/mol) 2(1)(19.716 kJ/mol) 2(3)(136.391 kJ/mol)
2.2 团簇Mo3S4的热力学稳定性
热力学稳定性能够判断生成的物质能否转化成其他物质,及是否存在自发反应的趋势,有助于研究化学反应过程。表1列出了团簇Mo3S4各优化构型的4种能量参数,包括校正能(EZPE),吉布斯自由能(G),吉布斯自由能变(ΔG)和结合能(EBE)。EBE=3EZPE(Mo)+4EZPE(S)-EZPE(Mo3S4),ΔG=G(Mo3S4)-3G(Mo)-4G(S)。依据能量最低原则,EZPE越小,EBE越大,构型就越稳定。
将新鲜荷叶置于恒温干燥箱中,60 ℃烘干至含水量8%以下,粉碎后过50目筛,密封于干燥自封袋中,置于干燥避光环境中保存。
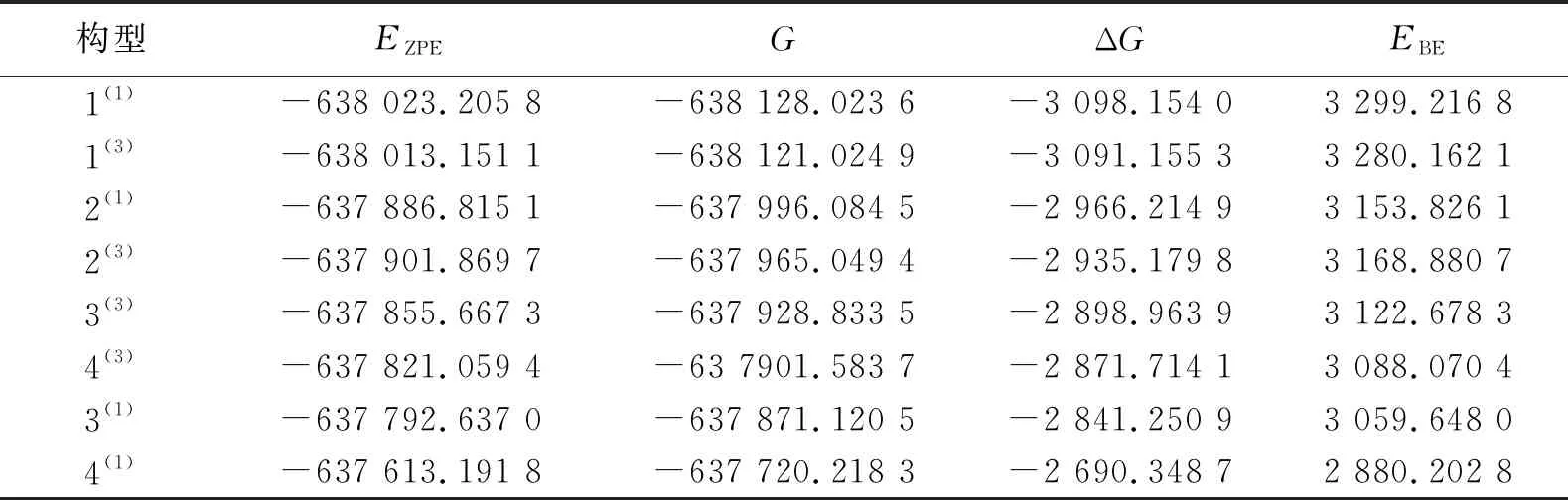
表 1 团簇Mo3S4各能量参数
从表1可知,构型1(1)的EZPE最小,EBE最大,即热力学稳定性最佳;反之,构型4(1)的热力学稳定性最差。其中EZPE(Mo)=-176 467.183 8 kJ·mol-1,EZPE(S)=-26 332.859 4 kJ·mol-1,G(Mo)=-176 510.416 7 kJ·mol-1,G(S)=-26 374.654 9 kJ·mol-1。
为了更好地研究团簇Mo3S4各构型的热力学稳定性,设定团簇的合成路径为:3Mo+4S→Mo3S4。EBE越大,构型中原子的结合程度越大,构型相对越稳定;ΔG小于零时,反应能够自发进行。各优化构型的ΔG均小于零(变化范围为-3 098.154 0~-2 690.348 7 kJ·mol-1)、EBE均大于零(变化范围为2 880.202 8~3 299.216 8 kJ·mol-1),说明单、三重态的所有优化构型均可自发形成且能够稳定存在。此外构型1(1)的EBE最大,ΔG最小,说明构型1(1)的稳定性最强。相反构型4(1)的稳定性最差。
2.3 团簇Mo3S4吸附氢原子性能
2.3.1 团簇Mo3S4优化构型的分子轨道 根据前线轨道理论[20],最高占据分子轨道(HOMO)与最低未占据分子轨道(LUMO)的HOMO-LUMO能隙值反映分子参与化学反应能力的强弱。团簇Mo3S4的第一步析氢反应是电子从该团簇的HOMO轨道转移到水分子的LUMO轨道,从而形成M—H结构(M为团簇Mo3S4)。团簇Mo3S4各稳定构型的α和β电子的HOMO轨道图以及水分子的LUMO轨道图如图2所示。其中阴影部分表示电子云密度较大的部分,阴影颜色表示电子的轨道波函数相位的正负,浅色表示正相位,深色表示负相位。轨道间存在自旋向上的α电子和自旋向下的β电子2种转移情况,因此要将各优化构型的HOMO轨道分为α-HOMO和β-HOMO进行讨论。
当原子相互接近时,同号轨道相互叠加,形成成键分子轨道,当电子进入成键轨道时可以形成更稳定的分子。从水分子的LUMO图可知,水分子周围基本都被深色包围,即波函数为负相位,说明水分子与团簇Mo3S4发生反应时,尽可能使团簇Mo3S4各构型的HOMO轨道中负相位电子与水分子重叠,同时保证正相位电子与其重叠部分为中心部分,才能更好地完成该团簇与水分子之间的电子转移。

(a) 1(1)α-HOMO (b) 1(1)β-HOMO (c) 1(3)α-HOMO
进一步分析图2可知在单重态中,构型1(1)与2(1)的β-HOMO图浅色阴影面积大于深色阴影面积,说明其轨道波函数相位为正的离域空间比轨道波函数相位为负的离域空间大。又因构型1(1)负相位的离域空间比构型2(1)小,这表明构型2(1)的HOMO轨道更容易与水分子的LUMO轨道重叠从而使构型吸附上氢原子。在三重态构型2(3)、3(3)、4(3)的β-HOMO图中,正、负相位的离域空间占比相差不大,说明其与水分子进行反应时α与β电子贡献程度相差不大。整体分析,构型1(3)的β-HOMO与2(1)的α-HOMO的负离域空间相较于其他构型更大,更易与水分子的LUMO轨道重叠而吸附上氢原子。由此可得,在团簇Mo3S4催化水分子析氢的吸氢反应中,构型1(3)与2(1)反应时较为活跃。
2.3.2 前线轨道能级差 由前线轨道理论可知,反应物之间能级差越小,越有利于促进电子在该团簇与水分子之间转移,使其更好地完成一系列反应。为进一步探究出吸附氢原子的最佳构型,表2列出了团簇Mo3S4的HOMO能级、水分子的LUMO能级及其差值,能级差ΔE=ELUMO-EHOMO)。
从表2可看出,构型1(1)的ΔE最大,构型4(3)的ΔE最小,说明在催化析氢的第一步反应中,构型4(3)R HOMO轨道中的电子最容易流入水分子的LUMO轨道,与氢原子成键生成Mo3S4—H;构型1(1)则最难发生该反应。构型3(1)与3(3)的ΔE相差不大,说明构型3(1)与3(3)在吸氢过程中表现出的反应活性相似。

表 2 团簇Mo3S4的HOMO轨道能级与H2O的LUMO轨道能级之差
2.4 (Mo3S4)—H的解吸过程

表 3 8种优化构型的HOMO能量与H2O的LUMO能级间的能级差
对比表2发现,当团簇模型变为(Mo3S4)—H 结构时,构型1(1)、2(1)和3(1)的ΔE值均增大,说明(1(1))—H、(2(1))—H和(3(1))—H与水反应时的活性降低,而其余M—H与水反应的活性增大。其中构型2(3)的ΔE增幅最大,构型3(1)的减幅最大。由表3中的ΔE可知,构型2(3)—H、4(3)—H的ΔE较小,说明构型2(3)—H与4(3)—H易发生解吸反应,其中构型4(3)—H的反应活性最强,其次是构型2(3)—H;构型1(1)—H在吸附氢原子后,其HOMO轨道的能级值最小且与水分子LUMO轨道间的能级差最大,因而在解吸过程中的反应活性最弱。结合2.3.2节分析结果可知,无论是在第一步吸附氢原子过程中还是在第二步电化学脱附过程中,构型4(3)的能级差均最小,所以相较于其他构型,构型4(3)更容易发生催化析氢反应,具有更好的催化反应活性。因此,在实际研发中应首先考虑依据构型4(3)设计催化剂Mo3S4的空间构型。
2.4.2 (Mo3S4)—H 结构模型的结合能 为进一步探究解吸过程中反应活性最优的结构模型,对各M—H模型的结合能进行比较,结果见表4。
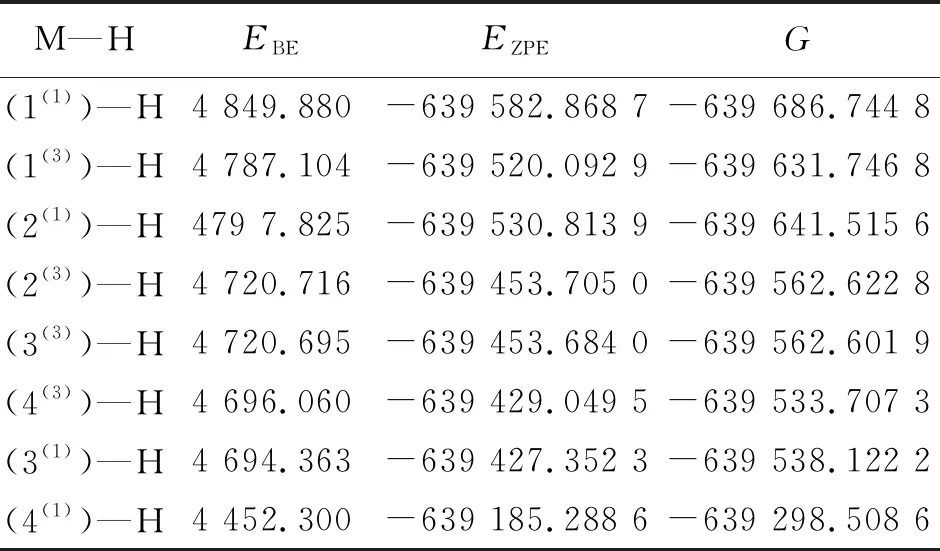
表 4 8种构型吸附氢原子后的结合能
M—H模型的结合能越小,越有助于解析过程的完成。结合表3数据不难发现,结合能较大的模型为(1(1))—H、(2(1))—H与(1(3))—H,说明这3个M—H模型的解吸过程不易完成,即不易与水分子进一步反应析出氢气。而仅分析结合能可知,构型4(1)的吸附模型结合能最小,说明(4(1))—H最易通过解吸反应析出氢气。
3 结 论
1) 团簇Mo3S4共存在8种优化构型,其中单、三重态各4种,各构型的热力学稳定性依次为:1(1)<1(3)<2(1)<2(3)<3(3)<4(3)<3(1)<4(1)。构型1(1)热力学稳定性最佳,构型4(1)热力学稳定性最差。
2) 构型1(1)的HOMO轨道给出电子进而更难与氢原子成键;构型4(3)的HOMO轨道中的电子最容易流入水分子的LUMO轨道,与氢原子成键生成Mo3S4—H;构型3(1)与3(3)的反应活性相似。在解吸过程中构型4(3)的HOMO轨道与水分子的LUMO轨道间的能级差最小,说明其反应活性最强。构型4(1)的吸附模型结合能最小,说明(4(1))—H最易通过解吸反应析出氢气。构型4(3)在团簇Mo3S4进行催化析氢反应时反应活性最佳。因此,在实际材料开发中应首先考虑依据构型4(3)设计出催化剂Mo3S4的空间构型,并对其展开相关研究。
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
科教新报(2021年11期)2021-05-12
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
科技传播(2019年22期)2020-01-14
中国科技纵横(2018年3期)2018-03-15
意林原创版(2017年11期)2017-12-01
中学物理·高中(2016年8期)2016-08-08
物理教学探讨(2009年7期)2009-06-08
数理化学习·高一二版(2009年1期)2009-03-19
