
浮选药剂与矿物表面吸附的空间匹配特性研究现状
2022-01-07 09:17:02刘文刚刘文宝彭祥玉
金属矿山 2021年12期
刘文刚 段 浩 刘文宝 彭祥玉
(东北大学资源与土木工程学院,辽宁 沈阳 110819)
浮选作为一项专门应用于矿物分选的技术,具有适应性强、分选效率高等优点,广泛应用于细粒和复杂物料的处理。在浮选过程中,浮选药剂(捕收剂、抑制剂等)起着不可或缺的作用,其在目的矿物表面的选择性吸附是实现矿物分离的先决条件。近年来,随着计算机技术与理论化学的发展,选矿研究者发现,选矿药剂,尤其是有机药剂,与矿物表面的相互作用存在一定的特异性匹配关系。即当有机分子的功能基团、电性、立体构型与矿物表面活性位点、电性、空间结构相互“契合”时,药剂分子才会在矿物表面发生特性吸附[1-3],如图1所示。
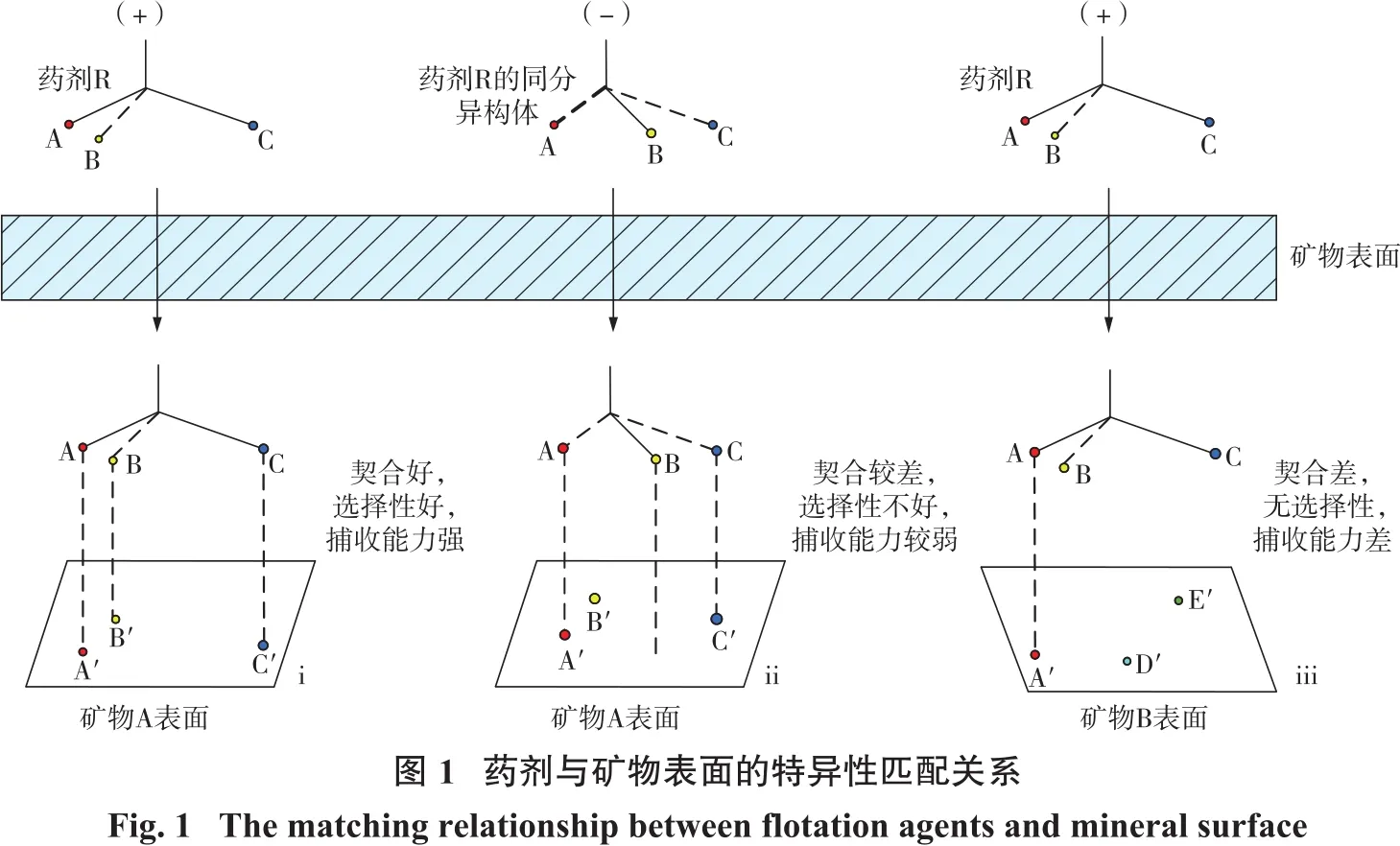
在浮选领域,目前关于药剂分子三维构型与矿物表面结构之间特异型契合的研究较少,厘清药剂分子与矿物表面的匹配关系有助于深入阐明药剂在矿物表面的吸附机理,提高浮选药剂设计过程的针对性与准确性,对开发高效的浮选药剂、推动浮选工艺的发展、提高我国资源的综合利用率具有极大的促进作用。因此,本文从同一药剂分子与矿物不同晶面的匹配关系、同一药剂分子与不同矿物表面的匹配关系和不同药剂分子与同一矿物表面的匹配关系3个方面出发,综述了近年来药剂与矿物表面空间匹配关系的研究成果,总结了其中的优势和不足,为进一步促进该方向的发展提供参考。
1 同一药剂分子与矿物不同晶面的匹配关系
在矿物的破碎磨矿过程中,矿物会暴露出不同的晶面,而不同晶面与同一药剂分子的作用形式存在差异,同一药剂分子与矿物不同晶面的匹配关系主要探讨了矿物表面的各向异性与其可浮性之间的关系。
锂辉石是金属锂的主要来源之一,其表面各向异性决定了锂辉石的浮选行为。XU、MOON、RAI等[4-6]研究表明,锂辉石的可浮性主要取决于(110)端面和(001)底面金属原子Al的性质。如图2所示,锂辉石(110)表面断裂两根Al—O键,形成带有一个单位正电荷的表面,每个Al的静电价为,-1价的羧酸根离子中2个O原子的静电价均为。因此,锂辉石(110)表面的金属位点Al的电性与羧酸根中O的电性能够很好匹配,且羧酸根基团尺寸也与锂辉石(110)表面Al原子所占面积相契合。而锂辉石(001)表面只有一根Al—O键断裂,表面静电价为,与-1价的羧酸根离子电性匹配程度较差。与此同时,锂辉石(001)表面上相邻Al原子之间的距离为0.632 nm,远大于羧酸根中O—O距离(0.219 nm),不利于羧酸根中2个O以双齿双核(2个O分别作用锂辉石表面2个Al原子)的形式与锂辉石匹配。

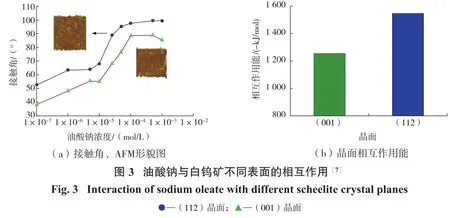
高志勇等[7]研究发现,相比于白钨矿(001)表面,油酸钠与白钨矿(112)表面作用的接触角更大,相互作用能也更低,结果如图3所示。通过对白钨矿晶体结构与油酸钠构型进行分析发现,在白钨矿(001)表面中,相邻活性位点Ca—Ca间距为0.524 nm,而油酸钠中O—O间距为0.219 nm,两者匹配性很差。因此,油酸钠在白钨矿(001)表面主要以单核双齿形式(羧酸根基团的2个O作用于白钨矿表面同一个Ca位点)吸附;而在白钨矿(112)表面,Ca—Ca间距为0.386 nm,O—O与Ca—Ca间距具有一定的匹配性,油酸钠以双核双齿形式(羧酸根2个O作用于白钨矿表面2个不同Ca位点)在白钨矿表面吸附。
另外,研究者发现羧酸根与方解石不同表面也存在着匹配关系。高志勇等[8]研究表明,方解石(104)和(110)表面 Ca—Ca间距均为 0.405 nm,羧酸根均以双核双齿形式吸附在2个表面,但(110)和(104)表面上钙原子的断键数分别是2和1,导致羧酸根与方解石(110)面匹配强度大于(104)表面。
2 同一药剂分子与不同矿物表面的匹配关系
同一药剂分子与不同矿物表面的匹配关系主要探讨了在矿物浮选过程中,药剂分子在目的矿物表面的选择性吸附机制。
锂辉石和长石同属铝硅酸盐矿物,具有相似的晶体结构和化学成分,但油酸钠与长石和锂辉石表面的作用具有明显差异。破碎过程中,长石(010)表面只断裂Na—O与Si—O键,未形成与油酸作用的活性位点Al,故油酸钠与长石(010)表面无吸附作用;长石(001)表面虽然会断裂一个单位的Al—O键,但活性位点Al—Al间距太大(1.152 nm),不利于油酸钠在长石表面的吸附。因此,油酸钠在长石表面的吸附性能偏弱。而对比锂辉石表面与油酸钠的吸附情况可知,油酸钠对锂辉石的捕收能力明显强于长石,相互作用能的计算也与浮选试验结果相对应,如图4所示[6,9-10]。

谭鑫等[11]研究了羟肟酸在白钨矿和萤石表面的吸附情况,研究结果表明,当羟肟酸在白钨矿表面以单核双齿形式(图5(a))吸附时,羟肟酸O位点与白钨矿表面Ca位点间距小于其与白钨矿表面O之间距离,即羟肟酸与白钨矿之间的静电引力作用强于静电斥力;相反,当羟肟酸在白钨矿表面以双核双齿(图5(b))构型吸附时,羟肟酸与白钨矿之间的静电斥力大于静电引力。这表明,羟肟酸阴离子与白钨矿表面呈双核双齿匹配相对较差,故羟肟酸倾向于以单核双齿形式在白钨矿表面吸附。而对于萤石,当羟肟酸以单核双齿形式吸附时(图5(c)),羟肟酸O位点与萤石表面Ca位点间距大于羟肟酸O位点与萤石表面F之间距离,表明此时羟肟酸与萤石表面的静电斥力大于静电引力,故羟肟酸易以双核双齿构型与萤石表面匹配(图5(d))。据此可实现高选择性浮选捕收剂的设计和开发。

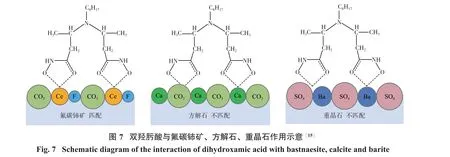
氟碳铈矿是轻稀土资源的主要来源,其伴生的脉石矿物为萤石、重晶石、方解石等。研究发现,氟碳铈矿(100)表面相邻Ce活性位点的间距分别为0.478 nm和0.706 nm,方解石(104)表面相邻Ca活性位点间距为0.405 nm和0.499 nm,而重晶石(001)表面相邻Ba活性位点间距为0.471 nm和0.560 nm,结果如图6所示。当药剂分子具有2个极性基团,且2个极性基团间距在0.700 nm左右时,药剂会与氟碳铈矿表面产生较强的吸附作用,而与方解石和重晶石匹配程度较差,从而提高药剂吸附的选择性[12-16]。基于这一理论,SUTTON等[14]结合量子化学计算与分子动力学模拟设计了系列双膦酸类捕收剂,并通过等温滴定量热分析验证了所设计的药剂与氟碳铈矿表面的吸附作用强于方解石表面。DUAN等[15-16]设计并合成新型双羟肟酸类捕收剂,如图7所示,该药剂与氟碳铈矿表面Ce位点匹配良好,而与方解石表面Ca位点和重晶石表面Ba位点匹配效果较差;单矿物浮选与人工混合矿分选试验均表明,所设计的药剂在氟碳铈矿、方解石、重晶石浮选分离体系中对氟碳铈矿具有较好的选择性,而直链结构的辛基羟肟酸易与氟碳铈矿、方解石、重晶石表面作用,导致其选择性较差。
GAO、DENG等[17-18]研究了羟肟酸与白钨矿和方解石表面作用时的匹配关系,如图8所示。研究表明,白钨矿表面钨酸根中O—O距离为0.290 nm,方解石表面碳酸根中O—O距离为0.222 nm,而羟肟酸离子中O—O距离为0.284 nm。羟肟酸离子中O—O距离与白钨矿表面O—O距离接近,所以与白钨矿表面具有更好的匹配特性,作用更稳定,导致白钨矿表现出更高的浮选回收率。
刘诚等[19]研究发现,多羧基类膦酸(PBTCA)中O—O距离(0.408 nm,0.497 nm)与方解石表面Ca—Ca距离(0.405n m,0.499 nm)接近,而与菱锌矿表面活性位点Zn—Zn间距(0.367 nm,0.465 nm)匹配性较差。因此,PBTCA在方解石与菱锌矿浮选分离过程中可以选择性抑制方解石。与此同时,该研究团队进一步考察了磷灰石与方解石分选过程中羟基亚乙基二膦酸(HEDP)对方解石的选择性抑制作用,研究表明,HEDP与方解石表面相匹配程度较高,而与磷灰石(211)表面匹配程度较差(Ca—Ca距离为0.781 nm)[20]。


陈建华等[21]通过密度泛函理论系统考察了黄铜矿与黄铁矿表面性质差异,研究结果表明黄铜矿表面Cu与硫胺酯药剂Z-200中S元素作用活性明显大于黄铁矿表面Fe与Z-200作用活性,因此Z-200与黄铜矿匹配程度优于其与黄铁矿表面,在铜硫分离中表现出较好的选择性。同时,该课题组研究了黄药与闪锌矿和铜活化后闪锌矿的作用差异,计算结果表明,在未活化闪锌矿表面,金属位点Zn被“包裹”于S元素中,较大的空间位阻阻碍了黄药中S与金属位点Zn的匹配作用,导致未经活化的闪锌矿在黄药浮选体系中几乎不可浮,而铜离子的活化显著降低了闪锌矿与黄药作用的能量壁垒。因此,经铜活化后的闪锌矿更易与黄药匹配,表现出了更好的可浮性[22]。
刘广义等[23]基于量子化学计算系统考察了4种硫代磷酸捕收剂的电荷分布信息(Mulliken电荷、自然电荷、前线轨道能量、偶极矩等)以及5种典型硫化矿表面金属位点的形成反馈键的能力(电子轨道分析、带隙等)。研究结果表明,4种捕收剂中DIBDTPI具有最强给电子能力与得电子能力,与Pb、Cu、Ag等硫化矿物形成反馈键能力较强,而与Fe与Zn的硫化矿物形成反馈键的能力较差。因此,DIBDTPI与Pb、Cu、Ag等硫化矿物电子匹配程度高于其与Fe或Zn的硫化矿物,对黄铁矿或闪锌矿表现出较差的捕收能力。
3 不同药剂分子与同一矿物表面的匹配关系
药剂分子的浮选性能与其分子结构密切相关,具备相似结构的药剂在浮选过程中可能会表现出较大差异的浮选结果,不同药剂分子与同一矿物表面的匹配关系主要讨论了药剂分子结构与其浮选行为之间的构效关系。
刘文刚等[24-28]设计并合成了几种具有多极性基团(羟基、胺基等)的新型阳离子捕收剂,并结合量子化学计算和分子动力学模拟,考察了捕收剂结构特征与其浮选行为之间的相互作用关系,结果表明,第二极性基团的引入降低了阳离子捕收剂的正电性,从而减弱了其与矿物之间的静电作用;同时,所设计的药剂分子极性基团尺寸较大,具有较大的空间位阻,削弱了药剂与矿物表面的匹配程度。陈攀等[29-31]考察了季铵盐极性基结构对高岭土浮选效果的影响,研究结果表明,当季铵盐头基部分为丁基时(RN+(C4H9)3,TTPC),丁基会提供更多活性位点与高岭土表面匹配形成CH…O氢键,从而提升了药剂在高岭土表面的吸附能力。
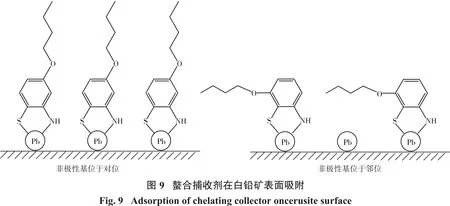
MARABINI等[32]考察了互为同分异构体的两种螯合类捕收剂在白铅矿表面的吸附作用,如图9所示,研究表明,当非极性基团位于极性基团对位时,药剂分子几乎可以与白铅矿表面所有Pb原子发生螯合作用,因而具有较好的捕收能力,而当非极性位于极性基团邻位时,由于空间位阻作用使药剂与矿物表面Pb的螯合作用减弱,导致其捕收能力有所下降。
王淀佐院士[33]考察了几种不同非极性基(戊基、异戊基、环己基、苯基)的二烃基二硫代次膦酸对毒砂和黄铁矿的浮选行为,研究结果表明,当非极性为直链结构时,毒砂与黄铁矿均具有较好的可浮性,而当非极性基为环己基与苯基时,由于非极性基团尺寸较大,会使药剂与矿物表面作用的空间位阻变大,造成药剂捕收能力的下降。
FILHO等[34]采用“药剂与矿物匹配指数”研究了两种多糖类抑制剂(淀粉与乙基纤维素)对羟基磷灰石和方解石的抑制作用,该指数反映了多糖类抑制剂分子中OH—OH距离与矿物Ca—Ca晶面距离之间的匹配度(图10),计算结果表明,淀粉与方解石的匹配指数大于乙基纤维素,表明淀粉对方解石抑制作用更强;对羟基磷灰石来说,淀粉与乙基纤维素的匹配指数均较低,表明两种多糖类抑制剂对羟基磷灰石抑制作用均较弱,后续单矿物浮选试也验证了这一结论的准确性。

现阶段,浮选药剂与矿物表面匹配关系的研究主要集中于以下几方面:①药剂与矿物表面不同晶面的匹配,即矿物表面各向异性与其可浮性之间相关关系研究;该方面的研究可深入指导磨矿过程,旨在通过选择性磨矿暴露出更易与药剂分子作用的矿物晶面,从而提高浮选效率。②同一药剂与不同矿物表面的匹配行为,即明确矿物表面活性位点间距与药剂分子官能团的契合度;该方面研究旨在厘清药剂在矿物表面的选择性吸附机制,为新型高选择性浮选药剂的设计开发提供指导。③不同药剂与同一矿物表面的匹配关系,即药剂分子结构与其浮选行为之间的构效关系研究;该方面的研究旨在考察不同结构的药剂分子与矿物表面的匹配关系,从而筛选出与矿物表面具有选择性吸附作用的药剂构型,以此提高药剂分子设计的针对性与准确性。
4 空间匹配关系在其他领域的体现
阻垢剂是指能够分散水中的难溶性无机盐,阻止或干扰难溶性无机盐(如碳酸钙、硫酸钡、氢氧化镁等)在金属表面的沉淀、结垢的一类药剂[35-38]。DAVEY等[39-42]探讨了多膦酸结构的阻垢剂中连接基团长度对重晶石结垢性能的影响,研究结果表明,当阻垢剂分子中膦酸基团间隔大于重晶石表面Ba—Ba距离(0.9 nm)时,阻垢剂连接基团(碳链)可以调整构型使两端的膦酸基团间距与重晶石表面Ba—Ba距离相匹配,达到近乎100%的阻垢率;而当阻垢剂分子中膦酸基团间隔小于0.9 nm时,只有一端的膦酸可以与重晶石表面Ba作用,阻垢效率只有50%左右。
REDDY等[43]研究了多羧酸类有机药剂在方解石阻垢方面的应用情况,结果表明,在羧酸根间距均为2个碳原子情况下,环羧酸环戊四羧酸(CPTCA)与四氢呋喃四羧酸(THFTCA)结构比较稳定,羧酸根间距几乎为固定值,可以稳定地与方解石表面匹配,其阻垢效果比直链羧酸三碳烯酸(TCBL)和柠檬酸(CTRC)更好;同时,相比于THFTCA的反式结构,CPTCA为顺式结构,分子内部4个羧酸基团均朝一个方向排布,能够提供多个位点与方解石相互匹配,因此CPTCA的阻垢效果好于THFTCA。
封盖剂可以与特定晶面匹配,使得晶体生长沿某一或某些特定方向,被广泛用于控制纳米材料的形貌[44-46]。BAKSHI等[47]对比了十二烷基三甲基溴化铵(DTAB)与双十二烷基二甲基溴化铵(12-0-12)作为封盖剂时对PbS晶体生长的影响,研究结果表明,当DTAB作为封盖剂时,DTAB会优先吸附于(111)表面,但也有部分吸附在(100)表面,因此形成的PbS纳米材料形状为星型;而当12-0-12作为封盖剂时,第二个月桂基的引入增加了药剂的空间位阻,使其无法与(111)表面匹配,但与(100)表面匹配良好,导致(100)表面被钝化,最终PbS晶体生长为立方体结构。
GRASES等[48]研究了多种羧酸在萤石晶体生长中的抑制作用,研究表明,对于含羟基的酒石酸和苹果酸,羟基O—羧酸根O距离为0.35~0.39 nm,与萤石表面Ca—Ca距离(0.380 nm)相匹配,且羧酸根O—O距离(0.285 nm)也与萤石表面F—F距离(0.270 nm)匹配,表现出较好的抑制效果;对于二元羧酸而言,草酸中首尾两端羧酸根中O—O距离与萤石表面F—F距离相匹配,丙二酸首尾两端羧酸根中O—O与萤石表面Ca—Ca距离匹配,丁二酸首尾两端羧酸根中O—O距离为0.490 nm,无法与萤石表面匹配;故丁二酸对萤石晶体生长几乎没有抑制作用。
5 结论与展望
随着计算机技术的发展以及人们对浮选药剂作用机理认识的深入,科研研究人员逐步意识到药剂分子结构与矿物表面之间的匹配关系决定了药剂分子的选择性吸附行为。基于前期的研究工作,研究人员初步明确了浮选药剂分子结构与矿物表面的空间匹配特性对其浮选性能的影响规律和作用机制。前期研究成果对选择性磨矿过程的调控以及浮选药剂选择性作用机制的分析具有很好的指导作用。然而,前期研究主要基于药剂分子的浮选行为来揭示药剂分子与矿物表面的匹配关系,在如何利用这种匹配关系针对性地开展高效浮选药剂设计方面进展较为缓慢。近年来,分子动力学模拟、量子化学计算等的快速发展,使人们在微观尺度解析浮选体系中复杂的化学反应,揭示药剂分子在矿物表面的吸附特征成为可能。因此,在后续研究过程中,应结合计算机模拟技术以及微观检测手段,进一步深入研究药剂分子与矿物表面的匹配特性,构建基于药剂与矿物表面空间匹配的高选择性浮选药剂设计方法,以期实现高精度与高准度的浮选药剂分子结构设计。
猜你喜欢
矿产综合利用(2023年5期)2023-10-31 02:32:28
矿产保护与利用(2021年4期)2021-10-26 02:17:56
矿冶(2020年4期)2020-08-21 08:16:24
新疆有色金属(2020年1期)2020-06-08 08:50:42
中国钨业(2019年2期)2019-10-21 09:30:02
中南大学学报(自然科学版)(2019年4期)2019-06-13 09:34:56
中国有色金属学报(2018年2期)2018-03-26 07:58:45
硅酸盐通报(2016年8期)2016-10-13 07:50:10
化工进展(2014年3期)2014-10-11 06:32:54
无机化学学报(2014年5期)2014-02-28 17:31:39
