
免疫细胞上TRPV4通道研究进展①
2022-01-05 05:40:16吴琼峰杜以梅
中国免疫学杂志 2021年22期
卢 凯 廖 杰 吴琼峰 杜以梅
(华中科技大学同济医学院附属协和医院心内科,武汉430022)
免疫细胞是免疫系统重要组成部分,参与多种炎症性疾病免疫应答过程,因此免疫细胞功能调节仍是免疫学研究重点[1]。Ca2+在众多免疫细胞内充当细胞外信号传递的第二信使[2]。免疫细胞在静止状态下维持较低的细胞内Ca2+浓度([Ca2+]i),而当其表面受体结合后可通过活化G蛋白或酪氨酸激酶级联激活磷脂酶Cγ1 等方式介导细胞外Ca2+内流[3]。Ca2+介导的信号传导对免疫细胞活化、增殖、分化、吞噬、分泌细胞因子及凋亡等功能十分重要。因此,调节免疫细胞内Ca2+信号可为免疫应答调控提供多个潜在靶点。
瞬时感受器电位离子通道香草素受体4(transient receptor potential vanilloid 4,TRPV4)是一种非选择性阳离子通道,在适宜温度(24~38℃)、低渗、缺氧、剪切力、内源性物质(花生四烯酸及其代谢产物EETs等)及外源性化学合成物(4α-PDD、GSK10167-90A 等)等刺激下激活后介导细胞外Ca2+内流[4]。TRPV4 活化已被证实参与多种炎症性疾病发生发展过程,而阻断TRPV4可能具有潜在抗炎效果。
多种免疫细胞上均证实有TRPV4 表达,随着各种免疫细胞上TRPV4 通道功能的阐明和各种TRPV4 相关药物的临床应用,TRPV4 有望成为免疫治疗药物的新靶点。本文对已有相关研究进行综述,旨在为TRPV4 作为免疫治疗靶点提供理论依据。
1 免疫细胞上TRPV4通道的激活机制
1.1 机械激活 免疫应答过程中,免疫细胞在多种炎症因子作用下发生迁移和黏附,免疫细胞受到剪切、牵拉、变形等机械刺激。免疫应答中机械力通过影响免疫细胞功能的新兴作用被称为机械免疫学[5]。TRPV4是细胞感受机械力变化的感受器之一,机械刺激可直接或间接激活免疫细胞上的TRPV4[5-6]。直接激活方式主要通过细胞膜脂质双分子层在变形过程中产生膜张力相关的能量差引起TRPV4 构象变化,进而促进其激活。而间接激活是指机械刺激通过细胞内信号传导级联激活TRPV4。这些信号级联主要包括机械敏感的黏着斑或紧密连接、细胞内机械敏感的第二信使、G蛋白偶联受体等,在其感受机械刺激后可触发TRPV4 激活或促进其向质膜转移。
1.2 免疫激活 炎症反应过程中产生的蛋白酶、5-羟色胺、组胺、TNF-α、IL-17 和蛋白酶激活受体2等可通过促进TRPV4 向质膜转移、磷酸化以及产生内源性TRPV4 激动剂等方式使免疫细胞上TRPV4表达增加或功能增强[6]。细菌表面的脂多糖(LPS)也被证实可直接激活TRPV4[7]。此外,多项研究表明革兰氏阳性菌及其产生的毒素(如肺炎球菌溶血素、α-溶血素、李斯特菌溶血素O 等)也参与TRPV4激活[8]。
1.3 温度激活 众所周知,感染引起的体温升高是免疫系统的重要激活剂。热敏离子通道可通过其温度依赖性激活参与温度对免疫细胞的调节,如温度依赖性STIM1 激活可通过诱导Ca2+大量内流参与并调节免疫功能相关基因表达。TRPV4 作为热敏离子通道,在体温变化范围内可被激活。由于TRPV4的热敏感性和Ca2+渗透性使其可作为免疫细胞感受体温变化的分子传感器,已有研究推测TRPV4 在先天免疫系统中可能参与热依赖性免疫调节[5,9]。
1.4 其他 此外,某些化学因素(如甲醛、过氧化物、酸、氯气等)、氧化的低密度脂蛋白(ox-LDL)等也被报道参与免疫细胞上TRPV4激活。
2 免疫细胞上TRPV4通道的表达与功能
2.1 TRPV4 通道与巨噬细胞 巨噬细胞包括定居于不同组织中的固有巨噬细胞(如肝脏中的库普弗细胞,中枢神经系统中的小胶质细胞、骨组织中的破骨细胞等)和游走于各种结缔组织中的巨噬细胞,可通过识别、吞噬和杀伤清除病原体等抗原性异物,也可作为专职抗原提呈细胞参与摄取、加工和提呈抗原过程引发适应性免疫应答。Ca2+依赖性信号传导参与调节巨噬细胞多种生物学功能,巨噬细胞钙稳态失调可导致多种疾病发生。
HAMANAKA 等[10]发现,机械通气产生的牵张力可激活肺泡巨噬细胞中TRPV4 参与呼吸机相关肺损伤。过度通气后伴随肺血管通透性升高,导致肺水肿,而TRPV4-/-小鼠这一现象明显减轻。将TRPV4+/+肺泡巨噬细胞过继转输至TRPV4-/-小鼠可使肺血管通透性再次升高,提示巨噬细胞上TRPV4活化参与介导过度通气引发的肺血管通透性升高。进一步研究发现,TRPV4 活化后巨噬细胞活性氧(ROS)和活性氮生成明显增加,迁移、黏附和吞噬功能增强,进而使肺泡通透性增加,参与肺水肿形成。随后,PAIRET 等[11]进一步研究证实,阻断TRPV4 可减少M1 型巨噬细胞促炎细胞因子分泌,改善气血屏障通透性。此外,RAYEES 等[12]和LI 等[13]发现,巨噬细胞TRPV4 参与调控LPS 诱导的肺泡巨噬细胞活化过程,TRPV4 介导的细胞外Ca2+内流通过CaN-NFATc3 途径使LPS 诱导巨噬细胞分泌IL-1β、TNF-α、IL-6等促炎细胞因子,阻断TRPV4则可通过减少炎症因子生成减轻肺组织炎症损伤,LI 等[13]研究还发现阻断TRPV4 后LPS 诱导的巨噬细胞ROS产生明显减少。表明肺泡巨噬细胞上TRPV4 活化后增强巨噬细胞介导的炎症和提高氧化应激水平导致肺血管通透性增高和肺部炎症反应加剧,而阻断TRPV4 则具有潜在治疗效果。但SCHERAGA等[14]研究发现,敲除TRPV4 使LPS 刺激的骨髓衍生巨噬细胞促炎细胞因子IL-1β 释放增加,而抑炎细胞因子IL-10 释放减少,与既往研究结论相反,提示不同来源巨噬细胞上TRPV4 的功能可能存在差异,此外,课题组还发现感染或纤维化肺组织中细胞外基质刚度增加触发巨噬细胞TRPV4 活化,使LPS 诱导的巨噬细胞吞噬作用增强,从而对细菌的清除能力增强。
研究发现,TRPV4活化是ox-LDL诱导的巨噬细胞泡沫细胞形成所必需的,阻断TRPV4 使基质刚度和LPS 刺激下巨噬细胞泡沫化减轻[15-16]。基质刚度增加或LPS 刺激下巨噬细胞上TRPV4 和CD36 共定位增加,巨噬细胞泡沫化加剧,阻断TRPV4 可减少巨噬细胞对ox-LDL 的摄取,减轻巨噬细胞泡沫化程度,提示TRPV4 可能通过影响CD36 功能参与调控泡沫细胞形成。因此,巨噬细胞TRPV4 有望成为动脉粥样硬化防治的新靶点。
破骨细胞是调节骨吸收的主要功能细胞,在骨发育、生长、修复、重建中具有重要作用。研究发现,TRPV4活化参与调控破骨细胞分化和活性[17-18]。破骨细胞中TRPV4 激活促进破骨细胞终末分化,增强吸收活性,导致骨质丢失,敲除TRPV4 可损伤破骨细胞骨吸收活性使骨量增加,主要由TRPV4 介导的Ca2+活化Ca2+/钙调蛋白复合物所介导。此外,破骨细胞还参与骨关节炎中软骨破坏、病理性骨重塑和滑膜炎症等病理过程。研究发现,阻断TRPV4 可加速骨关节炎形成,而激活TRPV4(如GSK1016790A或4α-PDD)可能有助于预防骨关节炎发生发展[19]。因此,破骨细胞上TRPV4 可能成为关节疾病新的治疗靶点。
小胶质细胞活化后参与中枢神经系统免疫应答,但激活的小胶质细胞同时分泌多种炎症细胞因子,加重脑组织损伤。LPS 可促进小胶质细胞活化和TNF-α 释放,KONNO 等[20]研究发现,这一过程可被TRPV4 激动剂4α-PDD 所抑制,提示TRPV4 激活参与减轻小胶质细胞活化和炎症反应。SHI等[21]却发现阻断小胶质细胞上TRPV4 可减少次声刺激下小胶质细胞IL-1β 和TNF-α 等促炎细胞因子释放,减轻神经元损伤。以上两种研究结果差异可能源于小胶质细胞上TRPV4 活化方式不同,导致其介导的效应存在差异。
LUO 等[22]发现,小鼠肠道肌层固有巨噬细胞上存在TRPV4功能性表达,TRPV4活化使巨噬细胞前列腺素E2 释放增加并以旁分泌方式作用于肠道平滑肌细胞,引起结肠收缩。而药物阻断TRPV4 或特异性敲除巨噬细胞TRPV4 后肠蠕动降低,并可逆转化疗引起的胃肠道动力过强,提示肠道巨噬细胞TRPV4 参与维持正常胃肠道运动,且靶向巨噬细胞TRPV4 有望成为胃肠道运动障碍性疾病治疗的新方向。此外,课题组还发现皮肤固有巨噬细胞中TRPV4 活化可通过下游血清素信号传导途径引发慢性瘙痒[23]。
SETH等[24]发现,肝脏中库普弗细胞上的TRPV4可感受氧化应激刺激,在其被氧化激活后可激活内皮eNOS 介导NO 释放,释放后的NO 可通过旁分泌的方式作用于临近肝细胞缓解肝细胞氧化损伤。因此,库普弗细胞上的TRPV4 参与肝脏内源性抗氧化防御机制。
综上所述,巨噬细胞中,TRPV4 在机械刺激、炎症因子等多种因素刺激下活化,其活化后介导Ca2+内流调控巨噬细胞ROS 形成、炎症因子释放及吞噬等功能,参与多种疾病病理生理过程。
2.2 TRPV4 通道与中性粒细胞 中性粒细胞(PMN)是固有免疫的重要组成部分。已有大量研究证实,Ca2+信号参与控制PMN吞噬、呼吸爆发等关键过程。SPINSANTI 等[25]首次在人PMN 中检测到TRPV4 mRNA 高表达。随后YIN 等[26]研究证实,小鼠PMN也存在TRPV4功能性表达,课题组通过酸诱导或氯气诱导肺损伤模型模拟胃酸诱导的吸入性肺损伤,基因敲除或药理学阻断TRPV4 后支气管灌洗液中蛋白减少,MPO 活性降低,IL-1β、IL-6、MCP等炎症因子释放减少,小鼠血氧饱和度明显改善,组织学损伤显著减轻。TRPV4-/-和WT 小鼠骨髓嵌合体模型发现,血细胞中TRPV4缺乏减少PMN向损伤肺实质浸润,提示阻断TRPV4可能通过调节PMN黏附和迁移减轻组织炎症。同时,阻断TRPV4 后抑制PMN迁移、趋化能力和ROS产生。
此外,ZHAO 等[27]和SOSTEGNI 等[28]研究表明,PMN 活化释放的弹性蛋白酶可通过激动蛋白酶激活受体2 活化腺苷酸环化酶、蛋白激酶A 和Rho 激酶等机制敏化TRPV4 引起炎症和疼痛。研究发现,TRPV4 激动剂可增加组织中PMN 浸润加重组织损伤,而阻断TRPV4则可减少组织中PMN浸润和炎症损伤[29-31]。提示TRPV4 活化与PMN 功能相关。但以上研究中,PMN 浸润减少是由于PMN 上TRPV4被阻断所致,还是由于阻断组织细胞上TRPV4 减少损伤间接导致PMN募集减少所致仍需进一步研究。
2.3 TRPV4 通道与肥大细胞 肥大细胞(mastocyte,MC)作为一种重要的免疫细胞参与机体过敏反应、慢性炎症、组织损伤修复、宿主免疫、肿瘤等多种疾病病理生理过程。MC 主要通过释放其胞浆颗粒内组胺、TNF、LT、PG、PAF 等多种生物活性物质发挥功能。持续的Ca2+内流是调节MC活化、趋化和脱颗粒的关键细胞内信号[32]。KIM 等[33]研究表明,人类MC 细胞系HMC-1 存在功能性TRPV4 表达。随后研究发现,剪切力、热等刺激可触发HMC-1细胞内Ca2+振荡频率提高并提高[Ca2+]i,而[Ca2+]i改变进一步驱动MC 脱颗粒,释放组胺等生物活性物质,提示MC 上TRPV4 可能参与这些因素驱动下的脱颗粒效应[34]。赘生型酒渣鼻发生与MC 密切相关,SULK 等[35]发现,赘生型酒渣鼻患者真皮TRPV4表达明显增加,进一步通过免疫荧光共定位技术发现TRPV4 增加与皮肤血管周围的MC 有关。随后,MASCARENHAS 等[36]发现,促酒渣鼻关键介质Cathelicidin 抗菌肽LL37可使人原代MC 中TRPV4表达呈剂量依赖性增加,该过程受G 蛋白偶联受体MrgprX2 活性调控。基因敲除或阻断剂HC-067047 阻断TRPV4后hMC脱颗粒能力明显下降,提示TRPV4参与调控MC 活化和脱颗粒,但具体机制尚不清楚,TRPV4 介导的Ca2+内流是直接作用于脱颗粒本身,还是作为信使促进MC 脱颗粒相关基因表达从而促进 脱 颗 粒 过 程 需 进 一 步 探 讨[37]。 此 外,SOLÍSLÓPEZ等[38]发现,小鼠腹膜MC也有TRPV4表达,激活IgE Fc 段受体,G 蛋白偶联受体MrgprX2 和内皮素1 后TRPV4-/-小鼠腹膜MC[Ca2+]i增加和脱颗粒等效应与野生型(WT)小鼠无明显差异。同时,TRPV4 激动剂GSK1016790A 未引起WT 小鼠腹膜MC[Ca2+]i升高或脱颗粒,与MASCARENHAS 等[36]研究结论不同,原因可能为人与小鼠物种差异、MC培养条件和MC类型不同。如TRPV1激动剂辣椒素可诱导小鼠骨髓来源MC[Ca2+]i升高和脱颗粒,但腹膜MC中却未观察到该现象。
MC 通过脱颗粒过程释放各种炎症介质(组胺、5-羟色胺、羧肽酶等)与哮喘发作密切相关[39]。多项研究发现TRPV4 活化参与哮喘发病过程。近期BONVINI 等[40]却研究发现原代人肺MC 上无TRPV4表达,哮喘发生主要与气道平滑肌细胞上TRPV4 功能增强间接触发MC 活化有关,但该研究结论需进一步验证。综上所述,目前MC 上TRPV4 的作用存在较多分歧,需要更多研究予以明确。
2.4 TRPV4 通 道 与 树 突 状 细 胞(dendritic cell,DC) DC 是专职抗原呈递细胞,可识别、摄取和加工外源性抗原并提呈抗原肽从而参与诱导适应性免疫应答和免疫耐受。大量研究表明,DC上存在多种Ca2+通道,这些通道参与调节DC 成熟、迁移和细胞因子分泌功能,如TRPV1 和CRAC 通道已被证实参 与 人DC 成 熟[41]。 多 项 研 究 证 实DC 中 存 在TRPV4 功能性表达[42-43]。最近,NAERT 等[43]发现,LPS刺激小鼠CD11c+骨髓衍生的DC(BMDC)成熟后TRPV4 表达和活性明显下调。同时,TRPV4-/-小鼠中CD11c+BMDC 对IgG 包被的微球内吞能力显著降低,提示TRPV4 参与Fc 受体介导的内吞过程,其机制可能为Fc 受体活化后增强吞噬膜附近TRPV4 活性和对机械应力的敏感性,而TRPV4 介导的Ca2+内流可促进肌动蛋白解聚,进而调控FcR 聚集和膜变形,参与受体介导的内吞过程。但缺乏TRPV4 不影响未包被微球内吞,提示TRPV4不参与非Fc受体依赖性内吞作用。 此外,TRPV4 缺乏似乎不影响CD11c+BMDC 分化、成熟和促炎细胞因子分泌等过程。DAVID 等[44]发现机械压力可增促进人DC 成熟和IL-12、IL-6、TNF-α、IFN-γ 等细胞因子分泌,但TRPV4是否参与该过程调控有待进一步研究。
2.5 TRPV4 通道与淋巴细胞 淋巴细胞在适应性免疫应答中占据核心地位。Ca2+参与调控淋巴细胞分化、代谢、抗体产生、细胞因子分泌和细胞毒性等多个过程,淋巴细胞内Ca2+信号异常可导致各种炎症、自身免疫性疾病和免疫缺陷综合征[45]。INADA等[46]首先在小鼠脾脏T、B 淋巴细胞亚群及小鼠T、B淋巴细胞系(BCL1 和EL4)中检测到TRPV4 mRNA表达。随后,MAJHI 等[9]证实,Jurkat 细胞(人T 细胞系)、人外周血T 细胞和鼠脾脏T 细胞上存在TRPV4功能性表达,且TRPV4 主要分布于细胞质及细胞膜。ConA 或TCR(α-CD3/CD28)(T 细胞激活剂)活化T 细胞后,TRPV4 蛋白表达显著增加。此外,RN1734 可抑制T 细胞活化和细胞因子IFN-γ、IL-2释放。而单独采用TRPV4激活剂4α-PDD 却未能活化T细胞。提示TRPV4在T细胞活化过程中可能仅起调节作用。RN1734 与TRPV1 抑制剂5'-IRTX 联用抑制T 细胞活化效果进一步增强,可能由于TRPV1 和TRPV4 可形成异源通道复合物,使2 个通道在T 细胞活化中产生协同作用和累加效应。总之,TRPV4 通道在T 细胞中功能性表达,并参与T 细胞活化过程。但TRPV4 通道如何在T 细胞中起作用及其如何与其他家族成员及其他通道间相互作用鲜有报道。未来将进一步探索TRPV4 在淋巴细胞发育过程中及在不同淋巴细胞亚群中的确切作用,以及明确淋巴细胞上TRPV4 在调节各种炎症性疾病过程中的作用。
3 结语和展望
综上所述,近年TRPV4 研究取得显著进展,多种免疫细胞上均证实存在TRPV4 功能性表达,且TRPV4 参与这些免疫细胞生物学功能调节(图1)。TRPV4 可直接介导细胞外Ca2+内流,也通过影响膜电位和Ca2+内向驱动力等因素间接参与Ca2+内流。此外,由于TRPV4 表达和活化方式不同,免疫细胞内Ca2+可呈现持续增高或钙振荡。不同细胞内Ca2+浓度和持续时间以及Ca2+振荡频率和幅度可能激活不同转录程序,参与免疫应答调节。
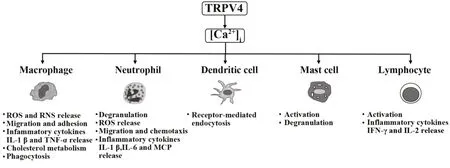
图1 免疫细胞上TRPV4通道表达与功能Fig.1 Expression and function of TRPV4 channels on immune cellsNote:ROS.Reactive oxygen species;RNS.Reactive nitrogen species.
越来越多研究关注到TRPV4 参与调节炎症性疾病发生发展,而免疫细胞上TRPV4 研究却仍处于初步阶段,且主要集中于经典免疫细胞上,而固有淋巴细胞、自然杀伤细胞、固有淋巴样细胞等免疫细胞是否存在TRPV4 表达和功能有待进一步研究。随着免疫细胞TRPV4 在炎症性疾病中确切作用和机制的进一步阐明,免疫细胞TRPV4 有望成为炎症性疾病治疗的新靶点。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10 09:15:38
现代临床医学(2021年4期)2021-07-31 07:55:54
作文成功之路·小学版(2020年6期)2020-07-27 01:48:28
哈尔滨医药(2016年3期)2016-12-01 03:58:33
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:37
癌变·畸变·突变(2016年3期)2016-02-27 06:15:25
医学研究杂志(2015年12期)2015-06-10 06:57:46
无机化学学报(2014年10期)2014-02-28 17:33:13
现代检验医学杂志(2014年1期)2014-02-06 01:29:19
西南军医(2014年1期)2014-02-03 03:06:31
