
ChIP-seq技术的研究进展*
2021-12-22 08:57:40施威扬袁明波
中国海洋大学学报(自然科学版) 2021年2期
施威扬,袁明波
(中国海洋大学海洋生命学院,山东 青岛 266003)
真核生物基因表达由RNA聚合酶、转录因子、组蛋白修饰等多种因素调控。为了在全基因组上定位DNA-蛋白质的互作区域,表观遗传学研究者提出了一系列技术方法。在1980年代,Gilmour D S等[1]和 Solomon M J等[2]最先提出了ChIP,用于富集和蛋白结合的DNA片段。在2000年代,基因组微阵列技术(Microarray)被用于定位这种富集的DNA,这一技术称为ChIP-chip[3]。随着二代测序技术的发展,ChIP-seq[4]技术在2007年首次被提出,并被很多大型跨国研究计划例如ENCODE(全称)广泛采用,从而成为研究DNA-蛋白质互作的金标准。但是ChIP技术对样本需求量大,存在着实验重复性差、信号低、背景高等缺点。此外,实验过程中容易引入DNA污染和DNA片段异质性,造成测序结果的假阳性。近年来,研究者对ChIP-seq技术不断进行改进:2011年ChIP-exo[5]技术被提出,利用外切酶作用于DNA片段,在一定程度上解决了ChIP-seq技术存在的假阳性、分辨率低、不能实现精确的映射(Map)等问题,实现了接近单碱基水平上定位DNA-蛋白结合位点。2013年,另一种表观组学技术ATAC-seq[6]出现,它利用Tn5转座酶处理细胞核中的染色质来研究染色质可及行区域,该技术常和其他方法结合被用于筛选可能的蛋白质结合位点。2017—2019年,一类全新的不依赖免疫沉淀的DNA-蛋白质互作研究方法,包括CUT&RUN[7-8]和CUT&Tag[9]彻底革新了DNA-蛋白质互作的研究模式。本文综述了ChIP-seq、CUT&RUN、CUT&Tag的技术原理,回顾了各项技术的重要应用场景,讨论了各技术存在的问题,并介绍了在单细胞水平研究DNA-蛋白质互作的技术挑战。本文最后对DNA-蛋白质互作研究技术的发展和应用进行了展望,为表观生物学研究提供借鉴和思路。
1 DNA-蛋白质互作技术的原理和应用
ChIP-seq、CUT&RUN和CUT&Tag是目前最常用的研究DNA-蛋白质互作的技术,其基本原理是先获得和目标蛋白质结合的DNA区域,然后对这一DNA区域进行高通量测序。通过这些技术揭示RNA聚合酶、转录因子结合位点和组蛋白修饰位点等表观遗传修饰信息,从而阐明基因开启或关闭的表观调控机制。例如:组蛋白第三亚基二十七号赖氨酸的三甲基化(H3K27me3)作为一种组蛋白甲基化修饰,标志着基因沉默;而组蛋白第三亚基四号赖氨酸的一甲基化(H3K4me1)和组蛋白第三亚基四号赖氨酸的二甲基化(H3K4me2)是活跃基因的标志,其富集区域容易进行转录和翻译[10]。表1给出了三种技术的实验流程比较。
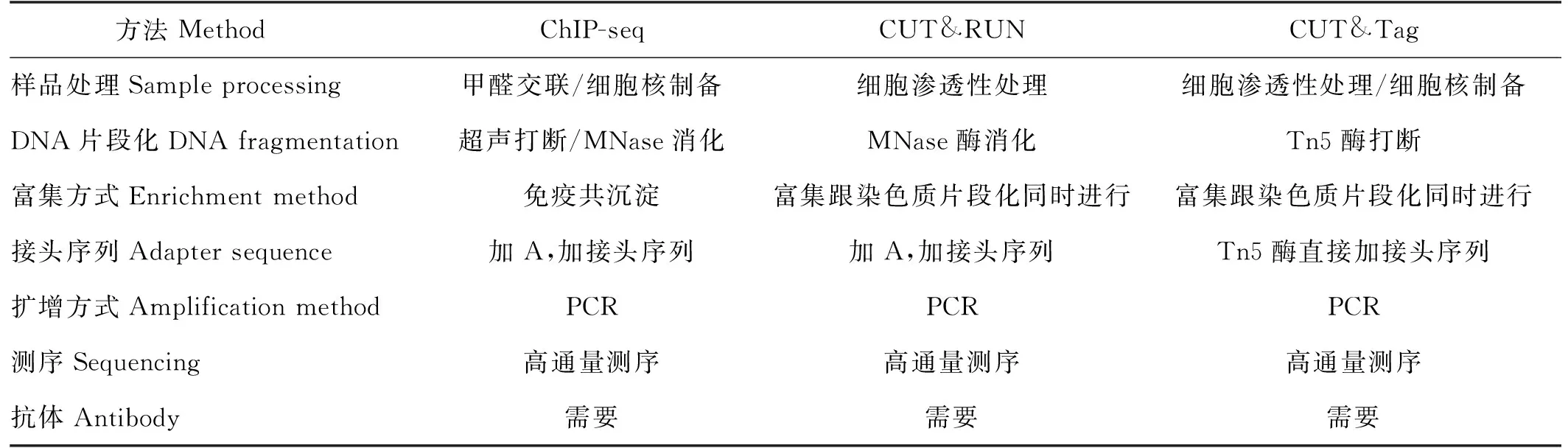
表1 三种技术的实验流程比较
1.1 ChIP-seq
ChIP-seq根据样品处理方式的不同可以分为X-ChIP-seq和Native ChIP-seq。X-ChIP-seq先利用甲醛处理样品,使DNA-蛋白质交联固定;然后裂解细胞,释放染色质并进行超声打断;之后用特异的抗体孵育,抗体结合到交联的DNA-蛋白质复合物上;再利用结合抗体的磁珠将DNA-蛋白质复合物沉淀富集;随后进行DNA与蛋白解交联,并纯化获得的DNA片段;最后将DNA片段补平,加腺嘌呤A,加接头序列(见图1A)。和需要交联的样本不同,Native ChIP-seq实验利用微球菌核酸酶MNase代替超声打断,直接对天然状态细胞中的染色质进行切割。MNase是一种同时具有内切酶活性和外切酶活性的核酸酶,主要作用于染色质上核小体之间的DNA区域。在Native ChIP-seq中,MNase消化未用甲醛固定的通透细胞或者细胞核,获得单个或多个核小体;之后同样通过抗体孵育和磁珠结合对DNA-蛋白质复合物进行富集;然后纯化获得的DNA片段通过之后的PCR扩增建库和高通量测序,获得与蛋白结合的DNA序列信息(见图1B)。
X-ChIP-seq需要进行甲醛交联、基因组超声打断、免疫共沉淀、DNA-蛋白质解交联以及文库构建加接头序列,这些繁琐的实验步骤容易造成样品的丢失。而Native ChIP-seq则避免了甲醛交联、超声打断、解交联这些步骤给样品带来的损害。这两种方法分别适用于不同性质的目标蛋白。例如:转录因子跟DNA通常是不稳定的瞬时结合,在Native ChIP-seq环境下很难捕捉,需要对样品进行甲醛固定;相反,组蛋白则是核小体组分,和DNA结合紧密,因此Native ChIP-seq常常用于组蛋白修饰位点的研究。但是,由于这两种方法都需要用经过免疫沉淀的步骤,用抗体和磁珠富集DNA-蛋白复合物,因此普遍存在样品需求量大,背景信号高,信噪比和分辨率低,以及容易出现假阳性的结果等缺陷。这些问题在一定程度上阻碍了ChIP-seq技术的应用。
尽管ChIP-seq技术存在种种弊端,其对表观基因组学的发展仍起到了极大的推动作用。ChIP-seq技术被广泛应用于转录、基因调控和表观遗传学领域。例如,Xiao R等[12],利用ChIP-seq技术对RNA结合蛋白(RNA-binding proteins, RBPs)结合位点分析发现RBPs主要分布在活性染色质区域,并在活性基因的启动子处有大量富集。之后通过与组蛋白修饰位点比较,发现RBPs结合位点与活性组蛋白修饰呈正相关,如:组蛋白第三亚基二十七号赖氨酸的乙酰化(H3K27ac);与抑制性组蛋白修饰呈负相关,如:组蛋白第三亚基九号赖氨酸的三甲基化(H3K9me3),这些证据表明RBPs能够调控基因的转录。其既可以直接调控转录,又可以通过和其他转录因子结合间接调控。作者进一步发现一个特定的RNA结合蛋白25(RNA binding protein 25, RBM25)可以和转录因子YY1相互作用并调控下游靶基因。YY1是一种广泛分布的转录因子,参与多种启动子的抑制或激活。ChIP-seq发现YY1既可以与RBM25共同结合到启动子区域,也可以单独结合,且YY1和RBM25共同结合比YY1单独结合能力更强,这表明RBM25的缺失会减弱YY1对下游靶基因的调控。这些结果显示RBPs能够广泛地与染色质直接或间接作用,从而实现RNA介导的转录调控,这一发现为研究转录调控的相关机理提供了新思路。Ran L等[13]用ChIP-seq对人类胃肠道间质瘤细胞系和前列腺癌细胞系中的转录因子ETS易位变体1(ETS translocation variant 1, ETV1)进行了全基因组定位,之后又在胃肠道间质瘤细胞系中对前叉箱蛋白F1(Forkhead box protein F1, FOXF1)进行了ChIP-seq分析,以确定FOXF1在胃肠道间质瘤细胞系的ETV1增强子位点的富集情况。同时,他们对H3K4me1和H3K4me3两个组蛋白修饰位点进行了ChIP-seq分析,并将其和ETV1富集进行了比较,发现FOXF1在增强子处与ETV1共域,并作为先锋因子发挥作用。FOXF1通过调节染色质可及性、增强子维持和ETV1结合,来调节依赖ETV1的胃肠道间质瘤细胞系的特异性转录组。这一研究为了解胃肠道间质瘤发生的分子机理提供了参考,并显示有望通过靶向干扰FOXF1表达来治疗胃肠道间质瘤。Cohen S等[14]在对蛋白SETX(Senataxin)在DNA双链断裂时的作用的研究中,用ChIP-seq对DNA双链断裂前后的SETX在全基因组定位进行了比对,发现在DIvA细胞中SETX被特异性招募到DNA双链断裂后活性转录基因上,且在诱导后表现出RNA:DNA杂交积累。由于SETX可以促进Rad51的募集,减少DNA末端的错误再连接,并在DNA双链断裂产生后维持细胞活力。这表明SETX在DNA双链断裂处通过限制移位以确保细胞存活,为SETX基因突变而引起的AOA2(常染色体隐性遗传性共济失调-眼运动不能2型)/ALS4(常染色体显性遗传性青年型)神经病变提供了新见解。Donaghey J等[15],用ChIP-seq技术对不同细胞中的前叉箱蛋白A2( Forkhead box protein A2, FOXA2)进行全基因组定位,发现FOXA2分布具有细胞特异性,获得了FOXA2所结合的基序(Motif)信息。为了探讨前表观遗传状态对先锋因子FOXA2, GATA4 和OCT4 结合的影响,作者定位了活性组蛋白修饰H3K27ac、H3K4me1和抑制性组蛋白修饰H3K27me3的分布,最后对G1停滞细胞中的FOXA2进行ChIP-seq分析。发现FOXA2占用和DNA可及性的改变可能发生在G1停滞细胞中。Wang C等[16]利用低输入样品量Native ChIP-seq(ULI-NChIP-seq),研究了哺乳动物胚胎发育过程中H3K9me3依赖性异染色质的重编程过程。通过小鼠胚胎着床前和着床后胚胎组织中H3K9me3的定位,绘制了小鼠早期胚胎中H3K9me3的全基因组分布图。发现H3K9me3在启动子和长末端重复序列(LTR)中表现出明显的动态特征,证明H3K9me3依赖性异染色质在早期胚胎发育过程中经历了戏剧性的重编程,为进一步探索早期胚胎的表观遗传机制提供了宝贵资源。因此,ChIP-seq技术已经成为表观生物学研究必不可缺的核心研究工具。
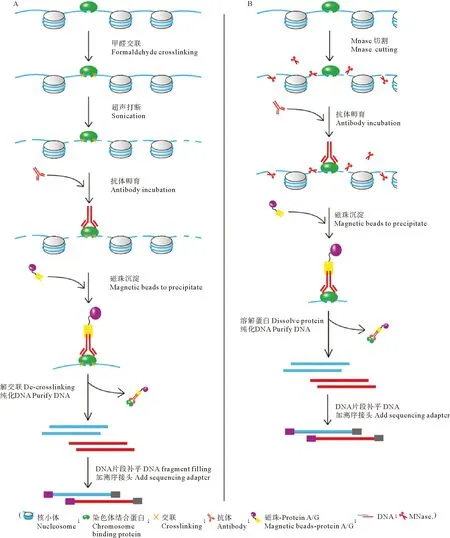
(A:交联染色体免疫共沉淀测序;B:天然染色体免疫共沉淀测序。A:Crosslinking chromation immunoprecipitation sequencing,X-ChIP-seq;B:Native chromation immunoprecipitation sequencing,Native ChIP-seq.)
1.2 CUT&RUN
CUT&RUN作为一种革命性技术,省略了免疫沉淀步骤,直接利用抗体把具有切割核小体间区能力的MNase酶靶向到原位,实现对蛋白质结合区域的核小体进行切割。该技术的核心是借助一种Protein A-MNase融合蛋白,Protein A是一种金黄色葡萄球菌细胞壁蛋白质,能特异地与人和哺乳动物抗体的Fc区(重链固定区)结合。其工作原理是:首先将细胞/核进行渗透处理,改变细胞通透性使得抗体和融合蛋白能够进入细胞核中;然后用目标蛋白的特异性抗体孵育,之后洗掉未结合的抗体,并与Protein A-MNase融合蛋白孵育,通过Protein A跟抗体的特异结合将MNase靶向到抗原周边。随后加入Ca2+激活MNase,MNase就会随机切割目标蛋白周围的DNA并从染色质上面释放出来。终止反应后,纯化这些释放出的DNA片段,然后加接头序列,建库测序(见图2)。由于只有被切割的DNA被释放并建库,而其余大部分DNA被留在细胞核当中,因此这一技术的背景非常低。
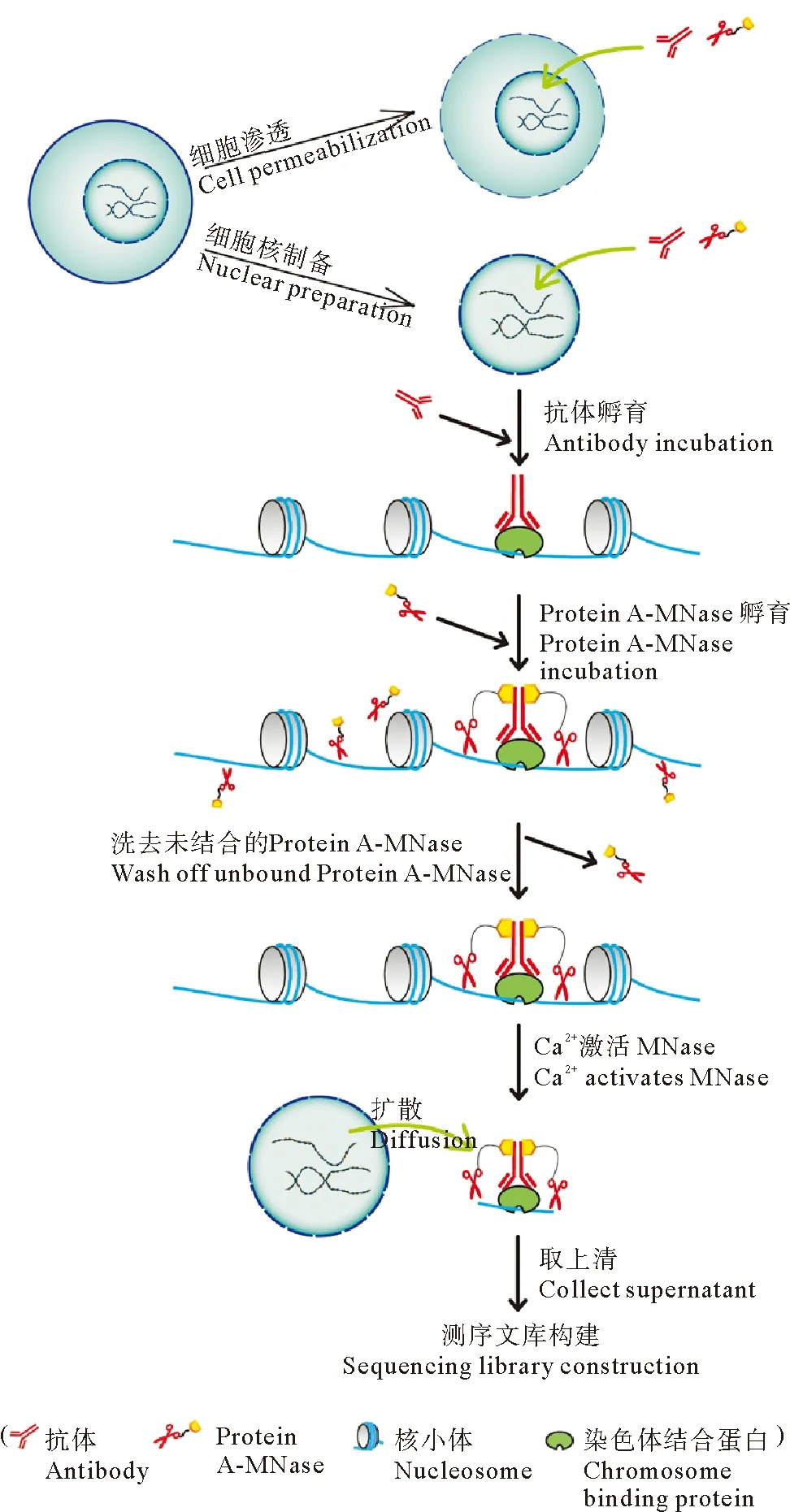
图2 CUT&RUN技术原理(参考Skene P J等[8])
CUT&RUN为表观生物学领域带来了重大革新。与X-ChIP-seq相比,CUT&RUN需要的细胞量大大减少,仅仅需要100个细胞就能定位组蛋白修饰位点;与Native ChIP-seq相比,CUT&RUN可以在低测序深度的情况下,就能得到高信噪比的数据。目前,CUT&RUN技术作为ChIP-seq的替代技术被广泛应用。Xia W等[17]利用CUT&RUN技术检测人类发育成熟的卵母细胞和早期胚胎中H3K4me3,H3K27me3以及H3K27ac的动态变化,揭示了人类早期发育过程中组蛋白修饰的重编程过程。研究发现,人类早期胚胎发育过程中的组蛋白重编程和小鼠相比呈现不同的动态变化。受精小鼠母源H3K27me3能够传递至囊胚,而人类H3K27me3在合子基因组激活前被大规模地去除,并在基因组激活后重新建立。在合子基因组激活前,H3K4me3分布在许多启动子区域以及基因远端开放区域,并伴随着这些区域的染色质开放性分布。此H3K4me3被称为预备H3K4me3(Priming H3K4me3),合子基因组激活后,这些区域会转变为激活或抑制的状态。这些发现为进一步了解人类早期胚胎的表观调控机制提供了重要依据。Zheng X Y等[18]利用CUT&RUN技术研究了拟南芥胚乳细胞的H3K27me3,获得了具有高灵敏度、特异性和可重复性的亲本特异性全基因组景观。这一研究进一步比较了用CUT&RUN和ChIP-seq获得的H3K27me3定位信息,发现二者富集峰重叠程度大,只在异染色质上存在差异,进一步证明了CUT&RUN可以基本替代ChIP-seq技术。CUT&RUN在临床研究中也发挥了重要作用。白血病是因为造血干细胞(HSCs)过度增殖,以及分化和凋亡受到抑制导致的非正常功能造血细胞大量积累并浸润其他组织,从而导致的机体造血功能障碍。IKAROS家族锌指2(IKAROS Family Zinc Finger 2, IKZF2)是一种染色质重塑剂,对染色质结构变化起到至关重要的作用。Park S等[19]用CUT&RUN对IKZF2定位分析,发现IKZF2在白血病干细胞(LSC)中高表达,其缺陷导致LSC功能缺陷。急性髓系白血病(AML)细胞中IKZF2的缺失可以减少集落形成,促进分化和凋亡,延缓白血病的发生。通过对LSC的基因表达分析、染色质可及性分析和IKZF2结合位点分析,表明IKZF2能够抑制分化转录因子C/EBP的表达,维持自我更新转录因子HOXA9和MYC的表达。后续功能验证实验证明IKZF2能调节AML-LSC程序,从而为通过靶向IKZF2来治疗髓系白血病提供了理论依据。最后,CUT&RUN技术被应用于胚胎造血过程的研究中。在胎儿向幼体的发育过程中,胎儿血红蛋白(HbF,α2γ2)的含量不断降低,成人血红蛋白(HbA,α2β2)比例不断升高。这一过程HbF沉默调节因子BCL11A起着关键作用。Macias-Trevino C等[20]利用CUT&RUN方法研究了从胎儿到成人血红蛋白转变过程中BCL11A所扮演的角色。首先用功能分析和蛋白质微阵列,确定了BCL11A的锌指簇和BCL11A的DNA结合序列(Motif)。还发现胚胎和胎儿时期珠蛋白启动子中存在这一Motif,特别是γ珠蛋白启动子中存在重复Motif。之后用CUT&RUN方法绘制红细胞中BCL11A蛋白合位点,发现BCL11A和γ珠蛋白基因启动子远端Motif上有结合。通过CRISPR编辑技术在 HUDEP-2细胞系中敲除这个远端Motif,可以阻止BCL11A结合,并导致γ珠蛋白基因启动子可及行增加。这一工作揭示了BCL11A对γ珠蛋白基因启动子的直接抑制是血红蛋白转换的基础,为深入了解人类胚胎发育过程中血红蛋白转变的分子机理做出了贡献。
1.3 CUT&Tag
在CUT&RUN技术中,MNase切割出的DNA需要纯化和建库,这一步骤中不可避免存在DNA的损失。在CUT&RUN技术基础上发展来的CUT&Tag则对此进行了进一步优化,它在靶向切割DNA的同时也标记了DNA,因此切割产物可以直接进行文库扩增。CUT&Tag的原理是用Tn5转座酶替代了pA-MNase融合蛋白中的MNase进行基因组的切割。Tn5转座酶可以催化双链进行重组交换[21],在打断DNA的同时在DNA片段两侧加入外源的扩增接头序列,因此它常被用于微量DNA建库测序[22]和染色质可及行分析[23-24]。CUT&Tag的技术原理是先用特异的一抗去孵育通透的细胞或者细胞核,洗去未结合的一抗;用组装好的Protein A-Tn5融合蛋白孵育,洗去多余的融合蛋白,形成Protein A-Tn5、抗体、靶蛋白和染色质复合物。之后,加入Mg2+激活Tn5转座酶,Tn5转座酶会将其结合区域邻近的DNA随机打断,并插入接头序列(见图3)。最后,直接经过DNA片段纯化和PCR扩增,便可以获得目标蛋白周边DNA区域的测序文库。
和ChIP-seq相比,CUT&Tag同样具有更高的信噪比;而跟CUT&RUN相比,它所需的细胞量更少,可以直接在单细胞水平进行检测。 Douse C H等[25]利用CUT&Tag技术成功研究人类沉默中枢复合体(HUSH)的亚单位TASOR。TASOR作为含有一个聚ADP核糖聚合酶(PARP)结构域的蛋白,对组装HUSH和抑制基因转座必不可少。可能由于TASOR分子大小或溶解度的问题,CUT&RUN技术并不适用于研究TASOR,此时CUT&Tag技术就显现出很大的优势。通过CUT&Tag对TASOR定位分析,发现TASOR富集峰和H3K9me3重合,表明TASOR对H3K9me3的富集至关重要。
2 单细胞水平的DNA-蛋白质互作
细胞是生物体基本的结构和功能单位,各细胞之间存在着异质性,因此在单细胞水平上研究细胞中各种组学信息至关重要。随着Drop-seq[26]、inDrop[27]和10×Genomics等单细胞转录组测序技术的出现,单细胞基因表达研究日渐成熟,单细胞多组学技术将成为未来研究的重点。其中,在单细胞水平上对DNA-蛋白质互作的研究已经获得了初步的成果。Rotem A等[28]提出Drop-ChIP,将液滴微流控技术和ChIP-seq联系起来,首次实现在单细胞水平上进行ChIP-seq。Drop-ChIP采用MNase代替超声打断,且在染色体免疫共沉淀之前就对单细胞染色体进行标记。通过对小鼠胚胎干细胞(ES),胚胎成纤维细胞(MEF)和造血祖细胞(EML)中H3K4me3和H3K4me2组蛋白修饰的定位分析,作者证明了Drop-ChIP技术可以用于区分单个细胞的异质性。CUT&RUN技术同样也被应用于单个细胞,Hainer S J等[29]通过对CUT&RUN实验条件进行了优化,改变了缓冲液、样品体积、抗体和融合蛋白孵育时间,以及文库制备和纯化方法,使CUT&RUN可以检测单个细胞中的DNA-蛋白相互作用信息。Ku W L等[30]提出scChIC-seq技术,除了采用和CUT&RUN一样的Protein A-MNase融合蛋白,其主要改进是相比CUT&RUN,它省略了分离目标DNA片段步骤,从而提高了DNA回收效率,使scChIC-seq技术适用于研究单细胞的DNA-蛋白质互作。同样,CUT&Tag技术也被应用于单细胞水平。 Wang Q等[31]等基于CUT&Tag原理提出了CoBATCH技术,通过组合标记(Combinatory labeling)的物理标记方式,用带有不同标记序列(Barcode)的Protein A-Tn5对细胞进行切割的同时使细胞带上第一轮标签;然后将所有细胞合并,重新分配到不同的孔,用不同的PCR引物进行扩增使细胞带上第二轮标签,从而实现一次数百个单细胞的检测。CoBATCH具有较高的信噪比,且每个细胞大约有100 00个读数,因此该技术可用于细胞种群异质性和亚型分析。

图3 CUT&Tag技术原理(参考Kaya-Okur H S等[9])
3 展望
DNA-蛋白质互作研究一直是表观遗传学领域的热点。作为研究DNA-蛋白相互作用的传统手段,ChIP-seq技术已经被广泛应用在动、植物研究中。在临床研究领域,ChIP-seq系统揭示了多种人类肿瘤中基因表达的表观调控机制,为肿瘤机制研究和治疗提供了重要生物学基础。而近年来发展出的CUT & RUN和CUT & Tag技术改变了传统的ChIP-seq研究模式,新的实验过程简单、实验条件容易控制,数据分辨率高,背景低,同时,这些技术对样品量要求低,可以直接进行单细胞水平的检测。尽管研究DNA-蛋白质互作的技术已经取得了很大进步,然而依然存在着不足。目前,最大的挑战是缺少合适的抗体来针对目标DNA-蛋白质互作区域。很多商用抗体质量不稳定,难以重复,而定制抗体生产周期长,成功率低。面对这一挑战,研究者提出了一些新的解决方法,例如直接在细胞中利用化学或生物修饰方法在蛋白上面加入标记,例如Branon T C[32]通过大肠杆菌生物素连接酶(BirA)融合蛋白在细胞内源蛋白上导入生物素(Biotin)修饰,从而可以用链霉亲和素来介导跟目标蛋白的特异结合。随着各种新的研究DNA-蛋白相互作用的技术的发展,以及各种对目标蛋白的化学和生物修饰方法的开发,整个表观遗传领域必将迎来新的发展。
猜你喜欢
畜牧兽医学报(2022年3期)2022-03-30 02:29:20
中国畜牧兽医(2022年1期)2022-02-15 10:46:40
河北果树(2021年4期)2021-12-02 01:14:50
今日农业(2021年11期)2021-08-13 08:53:24
上海公路(2019年3期)2019-11-25 07:39:28
现代泌尿外科杂志(2019年10期)2019-10-31 07:31:54
生物学通报(2019年2期)2019-06-15 01:33:42
福建基础教育研究(2019年10期)2019-05-28 08:27:04
遗传(2014年3期)2014-02-28 20:58:52
遗传(2014年3期)2014-02-28 20:58:49
