
2氰基吡啶-3噁二唑丙酸-稀土Gd配合物的合成研究
2021-12-22 06:59:20郭逸辰刘佳慧张诗齐
辽宁丝绸 2021年4期
郭逸辰 刘佳慧 李 静 张诗齐 张 芳
(辽东学院化工与机械学院,辽宁 丹东 118003)
1 实验
1.1 主要实验药品
2-氰基吡啶、盐酸羟胺、丁二酸酐均为化学纯试剂;Gd(NO3)3、无水碳酸钠、氯化锌、乙醇、甲醇、乙酸乙酯等均为分析纯试剂,上述药品均购自国药集团化学试剂有限公司。
1.2 主要实验仪器
AB-204S电子天平,珠海天创仪器有限公司;Spectrum 100型红外光谱仪,美国PE公司;ZF-20D紫外分析仪,巩义市予华仪器有限责任公司;旋转蒸发仪,杭州振和科学仪器有限公司。TGA-1000A热重分析仪,河南易棂洸有限公司。
1.3 合成方法
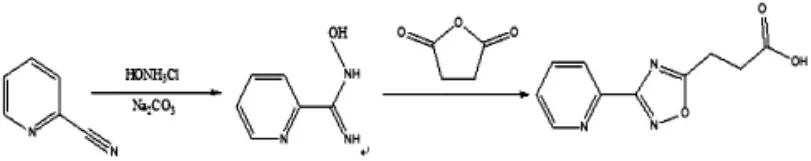
图1 2氰基吡啶——3噁二唑丙酸配体的合成
2 实验步骤
2.1 2氰基吡啶——3噁二唑丙酸的合成
准确称量2-氰基吡啶(20.79 g0.2 mol),溶于20 ml无水乙醇中,搅拌使其溶解至三口烧瓶中,盐酸羟胺(27.83g 0.4 mol)溶于40 ml蒸馏水中,超声震荡溶解后倒入三口烧瓶,滴加碳酸钠溶液,水浴升温至85℃,回流、恒温反应5h,反应终止静置、降至室温。经旋转蒸发,有白色针状晶体生成,过滤、水洗、结晶、烘干,得8.4g 2氰基吡啶偕胺肟。
准确称量2-氰基吡啶偕胺肟5.84 g(0.04 mol),丁二酸酐8 g(0.08 mol),混合后放入研钵中研磨,后将药品放入100ml单口瓶中,搅拌回流升温至155℃时,反应5 h,反应终止静置、降至室温。得固体,加入甲醇超声震荡;滴加氢氧化钠溶液使固体溶解,滴加盐酸有固体析出,即2氰基吡啶——3噁二唑丙酸配体,水洗、过滤、重结晶、干燥得到6.85 g固体。
2.2 稀土Gd配合物1的合成
准确称量2氰基吡啶-3噁二唑丙酸0.35g,Gd(NO3)30.27g,混合放入单口瓶中,并向单口瓶中加入30ml甲醇,5ml蒸馏水使之溶解。滴加氨水,常温反应8h,静置过夜,水洗、过滤、干燥,得稀土Gd配合物1。
2.3 稀土Gd配合物2的合成
准确称量2氰基吡啶-3噁二唑丙酸0.35g,ZnCl20.08 g混合放入单口瓶中,并向单口瓶中加入30ml甲醇,5ml蒸馏水,反应1h后加入Gd(NO3)30.27g,滴加氨水,常温反应8h,静置过夜,水洗、过滤、干燥,得稀土Gd配合物2。
3 结果与讨论
3.1 红外谱图分析
傅里叶变换红外光谱数据用Spectrum 100光谱仪,采用KBr压片,在4,000-500 cm-1采集。图中2氰基吡啶——3噁二唑丙酸配体在3400 cm-1左右有两个尖而窄的吸收峰,这是羧酸单体O-H的典型吸收峰;稀土Gd配合物1及稀土Gd配合物2的红外光谱,两个产物在3400cm-1处的吸收峰由于生成稀土配合物而变宽,且比配体发生了红移,稀土Gd配合物1比稀土Gd配合物2的红移波数下降,由此,证明生成了两个不同的稀土配合物。
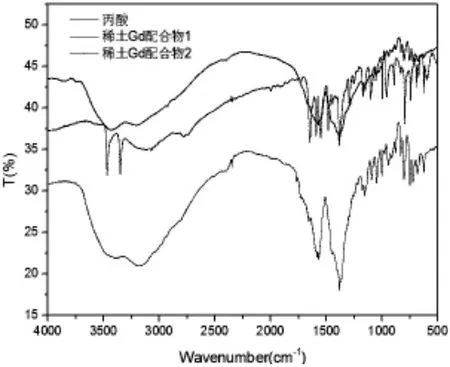
图2 丙酸配体、稀土配合物1及稀土配合物2红外谱图
3.2 紫外光谱分析
紫外光谱数据用ZF-20D光谱仪采集,波长在200-400nm。
由紫外谱图3可知1是丙酸配体紫外光谱,有2个吸收峰,分别在213、284处,2是稀土元素Gd配合物1紫外谱图,有2个明显吸收峰,分别在209、264处,3是稀土元素Gd配合物2紫外谱图,分别在217、265处有2个吸收峰,比较配体,2个配合物谱图形状发生变化。配合物紫外吸收光谱与配体基本相似,其峰都可以归属于配体内部的π→π*跃迁。但相对于配体,配合物1的两个吸收峰位置均有不同程度的蓝移;配合物2在短波处的吸收峰发生红移,在长波处发生蓝移;同时,配合物的吸收强度均在不同程度上小于配体。这主要是因为配合物产生后,π电子离域化程度减弱,π-π*跃迁能级差减小,从而使峰位蓝移,吸收强度减弱。
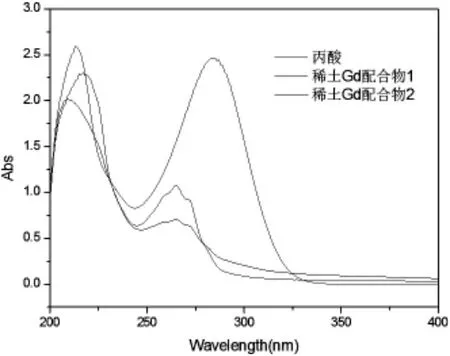
图3 丙酸配体、稀土配合物1及稀土配合物2紫外谱图
3.3 热失重谱图分析
热失重数据由TGA-1000A热重分析仪采集,温度在20-800℃。
由图4可得到,稀土金属配合物1在20-200℃之间质量保持稳定,到250℃时质量陡然下降,碳骨架解体,315℃后随温度升高,质量变化不明显,趋于稳定。此物质的质量下降说明配合物的分解。稀土金属配合物2热失重谱图同上。
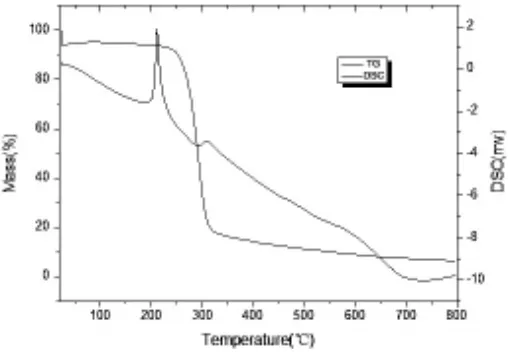
图4 稀土配合物1热失重谱图
3.4 荧光光谱
由图5可知丙酸配体、稀土配合物1及稀土配合物2有弱的荧光性,且稀土配合物的发光波长较丙酸配体红移。
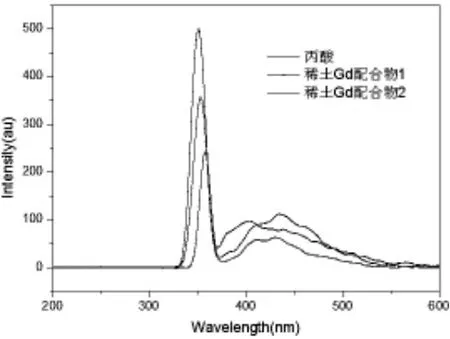
图5 丙酸配体、稀土配合物1及稀土配合物2荧光谱图
4 结论
以2-氰基吡啶、盐酸羟胺为起始原料反应得2氰基吡啶偕胺肟,再与丁二酸酐油浴熔融反应得到2氰基吡啶——3噁二唑丙酸,丙酸配体与稀土元素Gd反应,得到2个稀土配合物。通过红外谱图、紫外谱图、热失重进行结构表征,生成了2个新的稀土配合物,经过荧光光谱研究,配丙酸配体、稀土配合物1及稀土配合物2有弱的荧光性能,且稀土配合物的发光波长较丙酸配体红移。
猜你喜欢
上海文化(文化研究)(2021年6期)2021-11-30 06:32:24
煤气与热力(2021年4期)2021-06-09 06:16:56
昆明医科大学学报(2021年2期)2021-03-29 07:42:14
四川警察学院学报(2019年6期)2019-12-28 07:20:06
贵州农业科学(2019年9期)2019-10-10 08:20:26
环球时报(2018-08-15)2018-08-15 04:28:45
三联生活周刊(2018年12期)2018-03-30 08:30:20
文化产业(2016年6期)2016-10-19 19:13:47
西安建筑科技大学学报(自然科学版)(2014年1期)2014-11-12 13:03:42
生物加工过程(2013年1期)2013-03-11 18:31:35
