
一种猪伪狂犬病病毒gE基因TaqMan荧光定量PCR检测方法的建立
2021-12-20 14:08:32任玉鹏王一丹
养猪 2021年6期
任玉鹏,纳 吉,向 华,向 萌,王一丹
(西南民族大学畜牧兽医学院,四川 成都 610041)
猪伪狂犬病是由猪伪狂犬病病毒(Pseudorabies Virus, PRV)引起的猪的一种急性接触性传染病。除猪外,犬、猫和其他多种家畜均为其易感动物。PRV对各年龄猪只均可引起发病,以引起呼吸困难、神经症状和繁殖障碍为特征,特别是导致母猪流产,仔猪感染死亡率可达100%,危害极为严重[1]。我国长期通过免疫接种、抗体监测等手段持续对PRV进行防控和净化,这使得猪伪狂犬病疫情在一定程度上得到控制。但是,近年来我国PRV免疫失败的案例层出不穷,具体表现为免疫PRV Bartha-k61基因缺失疫苗的猪群再次出现感染,并且其发病率和死亡率呈现明显上升趋势,给我国PRV的防控带来新的挑战[2]。
对当前流行的PRV毒株分子特征研究结果表明,流行株基因组出现了不同程度的遗传变异,特别是自2011年后,PRV变异株的出现导致其在国内流行趋势逐步上升。近年来,在四川、河南、湖北和广东等多地均有PRV流行的报道[3]。对PRV变异株主要毒力基因测序分析发现,变异株与经典毒株处于不同的分支,经典毒株属于基因1型,变异株多为基因2型,在gB、gC、gD和gE基因均存在一定程度的遗传变异[4-5]。如gB基因的遗传变异多分布于其重要抗原表位(59~279氨基酸)区域,这可能是导致毒株部分抗原性改变的重要原因[6-7]。此外,高映雪等[8]对2013—2018年国内流行的64个毒株测序分析发现,所有毒株与经典株相比在gE基因均出现不同程度的变异,特别是第48位氨基酸处均存在1个天冬氨酸(D)插入。由于gE基因是PRV重要的毒力基因,上述氨基酸变异是否对病毒毒力产生影响值得进一步研究。同时,gE基因作为PRV重要分子检测靶点,是野毒株与基因缺失疫苗株的重要鉴别基因,该基因的核苷酸变化可能会导致部分已有检测方法的灵敏性下降[9]。所以,新的PRV分子检测方法有必要建立。
本研究拟建立一种基于TaqMan荧光定量PCR技术的PRV分子检测方法,以PRV的gE基因作为靶向基因设计特异性引物和探针,达到有针对性检测PRV野毒株的目的。同时对该方法的反应体系及条件进行优化,评估其敏感性、特异性和稳定性,并比较其与商品化试剂盒检测符合率,以期为PRV野毒株的快速检测提供可靠手段。
1 材料与方法
1.1 菌毒株及样本
试验于2020年7—12月在西南民族大学畜牧兽医学院动物医学实验室进行。PRV Bartha-k61疫苗株购自某实业股份有限公司;猪流行性腹泻病毒(PEDV)、猪细小病毒(PPV)、猪A群轮状病毒(PoRV)、猪德尔塔冠状病毒(PDCoV)、猪传染性胃肠炎病毒(TGEV)、猪链球菌(S.suis)、猪源大肠杆菌(E.coli)、猪源巴氏杆菌(P.multocida)、猪霍乱沙门氏菌(S.choleraesuis)和葡萄球菌(S.aureus)等用于特异性检验的菌毒株及核酸样本信息见表1;用于符合率检测的50份PRV已知阳性样本核酸及40份已知阴性样本核酸由西南民族大学动物医学实验室保存。
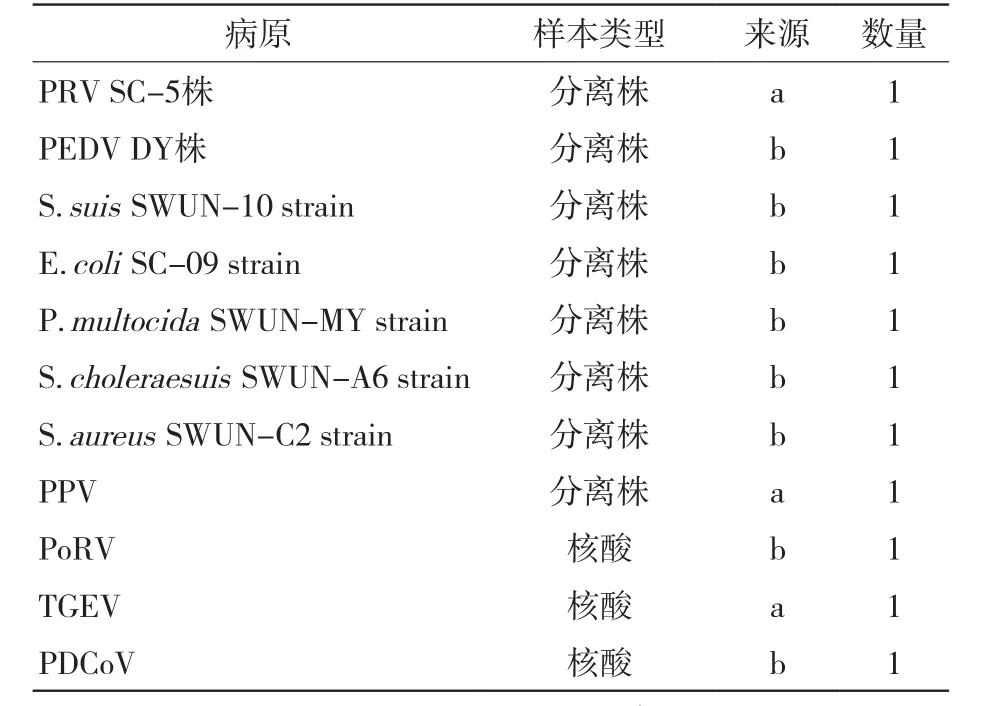
表1 菌毒株信息及来源
1.2 主要试剂
TIANamp Virus DNA/RNA kit和TIANamp Bacteria DNA kit购自天根生物科技(北京)有限公司;猪伪狂犬病病毒(gE基因)实时荧光PCR检测试剂盒购自北京世纪元亨动物防控技术有限公司;Premix Ex TaqTM(Probe qPCR)、BEST Agarose Gel DNA Extraction Kit Ver 4.0购自宝生物工程(大连)有限公司。
1.3 引物及探针的设计
用Maga 7.0对NCBI上登录的多个PRV毒株gE基因序列(KJ789182.1、MN240565.1、MK622299.1、MH507059.1、KU962917和KX170935.1)进行比较,通过对比分析找到gE基因的保守区域,采用Primer 5.0设计PRV gE基因长片段(562 bp)扩增引物(P1/P2)用于构建质粒标准品;进一步在该区域内用Beacon Designer 8设计用于检测方法建立的特异性引物(P3/P4)及探针(表2)。上述引物序列均由生工生物工程(上海)股份有限公司合成。

表2 PRV扩增引物
1.4 核酸提取
将病毒液和菌液各取200 μL,按照TIA Namp Virus DNA/RNA kit和TIANamp Bacteria DNA kit的操作指南,对包括PRV SC-5株(Vero细胞毒)在内的所有毒株菌核酸进行提取,并将抽取的样品放置在-80 ℃下保存。
1.5 质粒标准品的构建
分别对PRV SC-5株核酸进行PCR扩增,反应总体系为50 μL,Premix Ex TaqTM25 μL、上下游引物(P1/P2)均为1 μL、模板2 μL,dd水补足至50 μL。反应条件:94 ℃ 2 min,(94 ℃ 30 s,57 ℃30 s,72 ℃ 35 s)循环30次,72 ℃ 10 min。将扩增产物进行琼脂糖凝胶电泳。
根据Mini BEST Agarose Gel DNA Extraction Kit Ver 4.0试剂盒说明书对目的片段进行回收纯化,然后根据pMD19-T载体说明书进行产物连接并转化至E.coliDH5α感受态细胞中。将重组菌涂布接种于含有Amp的LB平板,取阳性克隆进行测序鉴定,将其命名为:pGEM-T-PRV。利用NanoDrop 2000测定质粒标准品的浓度并根据公式拷贝数(copies/μL)=6.02×1023(copies/mol)×浓度(ng/μL)/MW(g/mol),MW为平均分子量,计算核酸标准品的拷贝数。
1.6 反应条件的优化
以步骤1.5中所获得的度为104copies/μL的pGEM-T-PRV标准品为模板,按照单一变量原则,进行荧光定量RT-PCR反应条件和体系的优化。在反应体系中加入引物P3/P4,使其终浓度分别为0.2、0.4、0.6、1 μmoL/L;加入探针使其终浓度分别为0.05、0.1、0.15、0.2、0.5 μmoL/L,根据扩增曲线和荧光强度值进行引物和探针浓度的优化。采用最佳引物和探针浓度,在退火温度分别为56、58、60、62 ℃下进行反应,最终确定最优体系和条件。
1.7 反应条件的优化
将pGEM-T-PRV标准品进行101~107倍的稀释,并分别作为模板进行TaqMan荧光定量RT-PCR检测,分别以各稀释度质粒标准品拷贝数的Log值和CT值作为标准曲线的X轴和Y轴以绘制标准曲线,确定线性关系。
1.8 特异性试验
分别用PRV Bartha-k61株、PEDV、TGEV、PoRV、PPV、PDCoV、S.suis、E.coli、P.multocida、S.choleraesuis和S.aureus等菌毒株核酸作为模板,以无菌水为空白对照,进行TaqMan荧光定量PCR反应评估检测方法的特异性。
1.9 敏感性试验
将已知浓度的pGEM-T-PRV标准品进行10倍梯度倍比稀释,以各稀释度标准品混合液为模板,按照最佳反应条件进行一步法多重TaqMan荧光定量RT-PCR反应,以评价该方法的灵敏性。
1.10 重复性试验
以步骤1.9中稀释至103、105和107拷贝数的pGEM-T-PRV为模版,进行3次TaqMan荧光定量RT-PCR反应,每次试验设置3次重复,以评价该方法组内和组间的重复性,并将试验结果进行统计学分析。
1.11 与商品化检测试剂盒比较检测符合率
用本研究建立的方法与PRV实时荧光PCR检测试剂盒进行比较。同时检测50份已知PRV野毒感染阳性样本和40份PRV阴性进行检测,通过2×2列联表法进行统计学分析计算符合率和Kappa值,评价该方法检测结果与商品化检测试剂盒的一致性。
2 结果
2.1 质粒标准品的鉴定
对pGEM-T-PRV重组质粒进行PCR鉴定,经琼脂糖凝胶电泳后,出现与预期大小相符的、大小约560 bp的目的条带(图1)。对重组质粒的测序并经Blast比对分析,结果与已登录参考PRV毒株gE基因序列同源性为100%,表明质粒标准品构建成功。
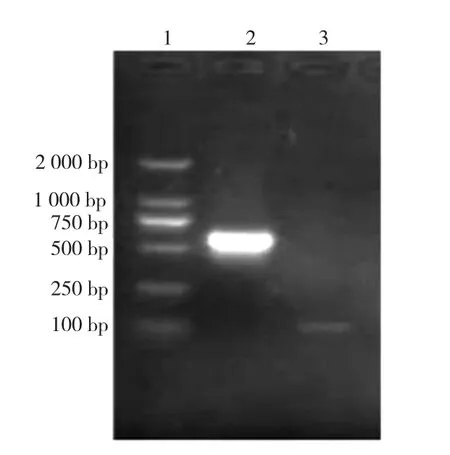
图1 PCR产物电泳结果
2.2 反应条件的优化
经优化结果表明,在PRV上下游引物浓度均为0.2 μmol/L,探针浓度为0.05 μmol/L、退火温度为58 ℃时,扩增曲线为S型光滑曲线,表明此为最佳反应体系和条件(图2)。
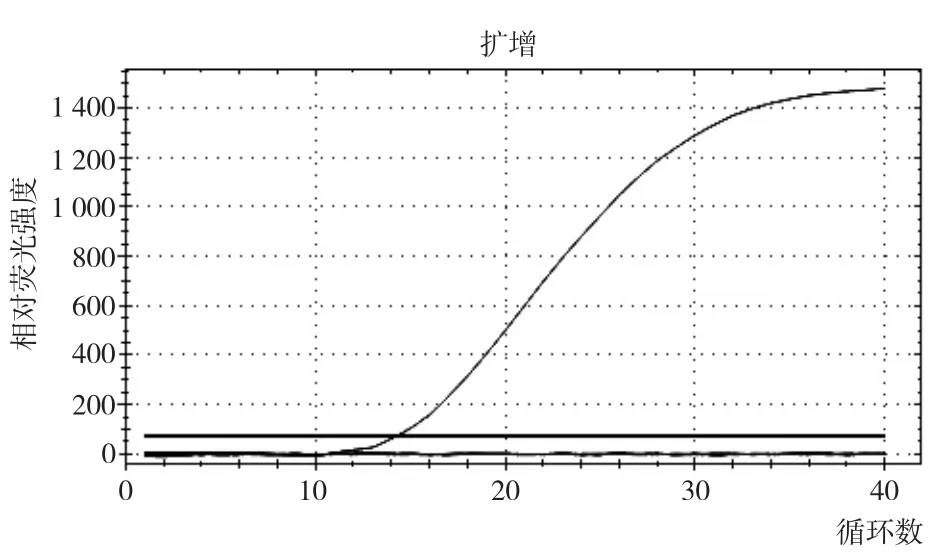
图2 优化后的荧光定量PCR检测结果
2.3 标准曲线的建立
以10倍倍比稀释的标准品浓度的Log值为X轴,以CT的值为Y轴绘制标准曲线(图3)。结果表明,该方法对标准品的扩增呈现良好的线性关系,其线性关系表达式分别为:y=-3.481 8 x+36.699,R2=0.995 5。

图3 荧光定量PCR的标准曲线
2.4 TaqMan荧光定量PCR的特异性
用本试验建立的PRV(gE)荧光定量PCR检测方法对PRV疫苗株、PEDV、TGEV、PDCoV、PoRV、PPV、S.suis、E.coli、P.multocida、S.choleraesuis和S.aureus进行检测,其CT值均≥38或无荧光信号,表明该方法具有良好的特异性。
2.5 TaqMan荧光定量PCR的检测下限
将PRV质粒标准品进行1 0倍梯度稀释(101~1011),然后进行TaqMan荧光定量PCR进行检测,结果显示PRV最低检测下限为3.92 copies/μL(图4),表明该方法具有良好的敏感性。

图4 荧光定量PCR法对10倍梯度稀释质粒的检测结果
2.6 重复性检测
重复性评估结果表明,无论是组内重复试验还是组间重复试验的变异系数(CV)均≤2%,表明该方法重复性和稳定性良好(表3)。

表3 PRV的重复性检测结果
2.7 符合率比较
用商品化的PRV(gE)荧光定量检测试剂盒与本研究构建TaqMan荧光定量PCR同时对50份PRV已知阳性样本核酸及40份已知阴性样本进行检测。结果表明,两种方法对所有样本的检测符合率为100% (k=1)。
3 讨论
传染病病原精准快速诊断对疫病监测预警和在早期防控过程中制定有针对性的政策措施具有重要意义。分子生物学诊断技术是实验室诊断中最常见和可靠的技术手段。其中PCR、荧光定量PCR和巢式PCR技术是世界动物卫生组织(OIE)推荐的非洲猪瘟、蓝耳病、猪瘟、伪狂犬病等猪重大疫病病原检测的常用技术[10]。而在所有的分子生物学检测技术中,荧光定量PCR具有高效、特异、灵敏、可定量、高通量、操作简便和无污染等一系列优点,在病原学检测中得以广泛应用。尤其是特异性TaqMan探针在荧光定量PCR中的应用,进一步提高了检测的特异性和灵敏度,适用于低拷贝数的临床样本检测[11]。因此,本研究采用探针法荧光定量PCR技术建立针对PRV野毒株的快速检测方法是其具有良好灵敏性和特异性前提和保证。此外,目前PRV基因结构约有近95% 已被定位和测序。PRV基因组编码近100种蛋白质。其中编码gE蛋白的基因是病毒复制的非必须毒力基因,其编码的囊膜蛋白质可促进感染细胞和临近感染细胞的膜融合,从而促进病毒的扩散,gE基因的缺失会导致病毒的嗜神经性毒力降低[12]。当前对PRV基因缺失疫苗的研究中,多采用确实gE、TK、gB等毒力基因的策略进行缺失株的构建。因此,对gE基因的检测可以实现只针对PRV野毒株的检测,而不对基因缺失疫苗株进行检出,可达到防止假阳性结果的目的。
本研究对PRV gE基因片段保守区域设计一对特异性的引物和探针,对反应体系和条件进行了充分优化,建立了一种检测PRV野毒株的TaqMan荧光定量PCR方法。结果表明,在上下游引物浓度分别为0.2 μmol/L、探针浓度为0.05 μmol/L、退火温度为58 ℃时扩增效果最佳,该方法检测下限可达到3.92 copies/μL,具有较高的敏感性。对包括PRV基因缺失疫苗在内的11种猪病相关的细菌和病毒株均无非特异性扩增,特异性良好。同时,该方法与商品化试剂盒同时检测已知阳性和阴性样本,结果符合率为100%,表明该方法检测结果可靠,可以初步用于临床样本的检测。本研究建立的PRV TaqMan荧光定量PCR方法可以为猪伪狂犬病的快速诊断和流行病学调查提供可供选择的工具。
猜你喜欢
科学大观园(2022年2期)2022-01-23 11:05:15
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:06
中成药(2018年12期)2018-12-29 12:25:44
中成药(2017年6期)2017-06-13 07:30:35
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
动物医学进展(2015年10期)2015-12-07 05:46:18
医学研究杂志(2015年4期)2015-06-10 06:42:43
物理实验(2015年9期)2015-02-28 17:36:47
特产研究(2014年4期)2014-04-10 12:54:12
