
响应面法优化制备蓝萼甲磺丁醚-β-环糊精包合物及体外活性研究
2021-12-15 02:11:18王娟,夏超
东南国防医药 2021年6期
王 娟,夏 超
0 引 言
冬凌草在民间处方中用作抗炎、解热和胃药[1-2]。蓝萼甲素A (glaucocalysin A,GLA)作为从冬凌草叶中提取的二萜类化合物[3],研究表明其具有抗菌、抗氧化、抗凝血和DNA保护活性[4-5]。GLA对癌细胞和肿瘤的生长也有不同程度的抑制作用[6]。由于GLA的疏水结构,水溶性较差[7],已成为临床抗肿瘤治疗的重要障碍。环糊精可以改善难溶性药物的溶解性,同时环糊精原料廉价易得,制备方法简单,易于操作,成为了改善药物溶解度的首选方法[8]。本研究通过环糊精制备成包合物来改善GLA的溶解度,从而发挥其抗肿瘤活性。
环糊精(cyclodextrin,CDS)是一类由α-1,4-连接的吡喃葡萄糖单元组成的晶体环状低聚糖[8-9]。近年来,它们作为有益的分子螯合剂广泛应用于食品、医药、化妆品等领域[9]。β-环糊精(β-cyclodextrin,β-CD)由于价格低廉,在其庞大的家族中使用最频繁,其空腔大小使其有可能包括大多数常见的客体化合物[9]。然而,β-CD水溶性差、溶血性强、肾毒性大,阻碍了其实际应用[10-11]。因此,羟丙基-β-环糊精(hydroxypropyl-β-cyclodextrin,HP-β-CD)和磺丁基醚β-环糊精(sulfobutyl ether β-cyclodextrin,SBE-β-CD)等人工修饰的β-CD应运而生[11]。修饰后的β-CD比未修饰的β-CD水溶性更好,毒性更小[12]。可能的原因如下:①SBE-β-CD中的磺酸基等取代基改变了β-CD表面的性质,从而提高了其水溶性。②随着修饰的β-CD分子量的增加,肾小管的重吸收减少,这会降低肾毒性。研究证实,SBE-β-CD在水溶性、毒性和增溶方面表现出比HP-β-CD更好的性能[12]。有研究报道制备了GLA与HP-β-CD的包合物,结果显示包合物后的水溶解度增加了13倍[13]。
本研究通过超声波法制备GLA在SBE-β-CD中的包合物,并对实验条件进行优化,以获得最高的包合效率。并研究GLA-SBE-β-CD对肿瘤生长的影响,采用MTT法检测GLA-SBE-β-CD对4种肿瘤细胞的体外抑制效果,为GLA在抗肿瘤的临床用药上提供有价值的剂型参考。
1 材料与方法
1.1 材料与仪器GLA(批号:20180908)由苏州大学药学院(中国江苏)提供。5-氟尿嘧啶购自TCI上海有限公司(中国上海)。SBE-β-CD(南京巨环医疗科技有限公司,平均相对分子质量Mw=2.24 kD,平均取代度为7),牛血清(上海洛神生物技术有限公司),胰蛋白(上海碧云天生物技术有限公司),甲基叔丁基醚(南京巨环医药科技有限公司),冬凌草甲素(中国药品生物制品检定所),3-(4,5-二甲基-2-噻唑基)-2,5-二苯基-2-H-四唑溴化物(Sigma-Aldrich Saint Louis, MO)。甲醇为色谱级,本实验中使用的水为超纯水。除非另有说明,所有其他材料均为分析级。细胞:人宫颈癌细胞Hela、人肺癌细胞A549、人肝癌细胞HepG2和人宫颈鳞癌细胞SiHA的细胞株均来自上海细胞所。液相色谱[(HPLC LC-20A,岛津(日本)仪器有限公司)],超声仪(上海科导超声仪器有限公司),冷冻干燥机(上海继谱电子科技有限公司),差示扫描仪(DSC,美国TA仪器有限公司),粉末X射线衍射(XRD, 北京新源志勤科技开发有限责任公司),扫描电子显微镜(S-4800, Hitachi, Tokyo, Japan),日立离子溅射机(E-1030, Hitachi, Japan) 。
1.2 色谱条件用配备LC-20A泵和紫外(PDA)检测器的岛津高效液相色谱测定GLA的浓度。色谱柱为Kromasil反相C18柱(4.6 mm×150 mm,5 μm)。流动相为蒸馏水∶甲醇(1∶1;V/V),流速为1.0 mL/min进行等量洗脱。柱温25 ℃,检测波长231 nm。
1.3 方法
1.3.1 制备GLA-SBE-β-CD包合物采用超声法和冻干法制备GLA-SBE-β-CD包合物[14-16]。将一定量的SBE-β-CD的水溶液在超声机中的烧瓶中恒温制备,然后在丙酮中慢慢滴加越来越多的GLA(GLA与SBE-β-CD的摩尔比在1∶5至1∶1之间)到不同的烧瓶中。所得溶液在超声下络合一段时间,直到达到平衡,然后在环境温度下放置2 h以冷却。之后,将冷却后的溶液在真空干燥器中冷冻干燥,通过0.45 μm膜过滤后,冷冻干燥得到固体复合物。准确称取一定量的GLA-SBE-β- CD,溶解于流动相中,将所得溶液约20 μL注入HPLC中,分析GLA的浓度。通过测定包合效率来选择得到的包合复合物。
1.3.2 GLA-SBE-β-CD制备条件优化在确定GLA与SBE-β-CD的摩尔比为1∶3为包合物制备的最佳组成基础上,考察SBE-β-CD水溶液的温度、时间、浓度等实验条件对包合效率的影响。因此,对这3个因素进行单因素检验,确定各因素的总体水平。然后采用响应面方法(RSM)等统计方法,通过对温度(A)、SBE-β-CD浓度(B)和包合时间(C)等操作因素的优化,使包合物的产量最大化,设定结果见表1。在本研究中根据试验设计结果,采用Box-Behnken设计(BBD)进行拟合,建立数学模型(Box-Benhnken设计的三因素三水平的响应面模型图。响应曲面图可以反映各变量与包合效率的关系:曲面越陡,响应变量对变量变化越敏感。等高线图的形状可以清楚地反映两个变量之间的交互作用是否显著:椭圆形等高线表示两个因素之间的交互作用显著,反之,等高线的圆度表示两个因素之间的交互作用不显著),且生成相应的多项式方程,便于计算GLA-SBE-β-CD的实际包封率(Υ),计算公式如下:

表1 Box-Benhnken检验的三因素和三水平正交实验设计表
Υ=β0+β1A+ β2B+ β3C+β4AB+β5AC + β6BC+ β7A2+β8B2+β9C2
其中Υ:响应变量即为包封率;β0:表示17个随机实验的所有定量结果的算术平均值的截距;β1-β9:根据Υ的观测实验值计算的系数;A、B和C:表示实验因子;AB、AC、BC:因子交互作用;A2、B2和C2:代表二次项。
1.3.3 GLA-SBE-β-CD增溶效果的研究将过量的GLA和GLA-SBE-β-CD分别加入10 mL蒸馏水中,然后放入恒温振荡器(25 ℃)中24 h,之后将得到的混合物通过0.45 μm膜过滤,并使用HPLC方法分别测定饱和溶液中的GLA的含量,计算其溶解度。
1.3.4 DSC分析差示扫描量热法(DSC)研究GLA-SBE-β-CD包合物(a)及其物理混合物(b)的热性能,并记录GLA(c)和SBE-β-CD(d)的DSC曲线,以验证GLA在SBE-β-CDs空腔中的包结或部分包结。使用差示热扫描仪对GLA、SBE-β-CD、物理混合物以及GLA-SBE-β-CD在氮气吹扫条件下进行了热分析。将3~5 mg样品放在坩埚上,加热温度为50~350 ℃,升温速率为10 ℃/min。
1.3.5 XRD表征在PW1720型X射线发生器和PW1710型衍射仪上研究GLA、SBE-β-CD、其物理混合物和GLA-SBE-β-CD的粉末X射线衍射图谱。工作电压为40 mA,电流为40 mA,由铜的Kα辐射产生。对GLA、SBE-β-CD、其物理混合物和GLA-SBE-β-CD粉末进行扫描,扫描步长为0.04°~35°,扫描速度为0.02°/s,用Jade6.0 XRD图形处理软件(Materials Data,Inc,Irvine,CA)对样品进行X射线分析。
1.3.6 SEM表征用扫描电子显微镜对GLA、SBE-β-CD和GLA-SBE-β-CD的表面形貌进行研究。在日立离子溅射机上,用双胶带将粉末样品粘在试样台面上,涂上导电铂膜,观察样品并记录照片。
1.3.7 人源癌细胞的抑制研究选用临床诊断中最为常见且致死率较高的4种人癌细胞,初步研究GLA-SBE-β-CD包合物和GLA对人源癌细胞的抑制效果。以含10%胎牛血清的RPMI-1640为培养基,采用梯度稀释法制备不同浓度的GLA和GLA-SBE-β-CD(1、10、100 μg/mL)。将4种对数生长的癌细胞悬液接种到96孔板中(细胞密度调整为1×104个/孔),分别加入不同浓度的GLA或GLA-SBE-β-CD。空白组不给予培养液。对照组只给予不含药的培养液。在37 ℃,5%CO2条件下培养24 h后,用10 μL MTT(3-[4,5-二甲基噻唑-2-基]-2,5-二苯基四氮唑溴化铵)和90 μL新鲜培养基取代RPMI-1640培养基,继续培养4 h,每孔加入100 μL二甲基亚砜(DMSO),在摇床中络合10 min,直至甲醛结晶完全溶解。用StatFAx-2100酶联免疫吸附仪在490 nm波长处检测二甲基亚砜-细胞混合液的吸光度,平行检测一式三份,取平均值计算癌细胞抑制率(IR):
IR=[1-(ODdrug-ODblank)/(ODcontrol-ODblank)]100%
其中ODdrug表示药物组的光密度,ODblank表示空白组的光密度,ODControl表示对照组的光密度。
2 结 果
2.1 GLA-SBE-β-CD包合物的制备随着SBE-β-CD的加入,包合效率提高,当GLA与SBE-β-CD的比例达到1∶3时,包合效率超过80%。当比例从1∶3继续增加时,包合效率未见明显提高。考虑到辅料、原料药的浪费和加工难度的增加,确定GLA与SBE的比例为1∶3为包合物的最佳比例。见表2。

表2 不同摩尔比下蓝萼甲素A在磺丁基醚β-环糊精中的包合率
2.2 Box-Behnken设计法优化GLA-SBE-β-CD制备条件实际拟合方程式为:
Υ=-173.65+9.42A+2.34B+0.974C+9.89×10-3AB-2.687×10-3AC-3.67×10-3BC-0.139A2-0.0685B2-0.011C2
BBD矩阵及响应值见表3。三维响应面和等高线图见图1。响应面二次模型的方差分析(ANOVA)显示见表4。结果表明,该模型具有较高的F值(33.34)和极低的P值(<0.0001),表明该模型具有统计学意义。“拟合不足”的P值为0.0666(>0.05),表明实际结果与拟合模型无显著性差异。R2和调整后的R2分别为0.9772和0.9479,表明实验因素与响应变量之间有很好的相关性。温度(A)、SBE-β-CD浓度(B)、包合时间(C)的F值分别为1.57、5.86、0.91,表明包合效率受SBE-β-CD浓度(B)的影响最大,受包合时间(C)的影响最小。变异系数(CV)为3.47%(<15%),结果均在正常标准范围内。由响应曲面图可见,SBE-β-CD(B)的浓度对包合效率的影响最大。由等高线图可见,AB和AC之间的交互作用是显著的,而BC之间的交互作用不显著。

图1 Box-Benhnken设计的三因素三水平的响应面模型图

表3 Box-Benhnken设计的三因素三水平矩阵拟合结果和响应值(R)
根据STATEASE公司的Design Expert软件(MN 55413,美国)的计算结果,可以得到最佳的实验参数和理论上的最大包合效率。结合实际试验条件,确定SBE-β-CD浓度为18.5%,包合温度为35 ℃,包合时间为42 min。在最佳工艺条件下进行制备,平均产率为87.28%,相对标准偏差(RSD)为1.02%。这一包合率略低于理论值(87.40%),但在误差范围内。
2.3 GLA-SBE-β-CD增溶效果的研究实验结果表明GLA的在水介质中的溶解度为0.213 mg/mL,而GLA-SBE-β-CD的溶解度为17.96 mg/mL,已经达到GLA的84.3倍,说明GLA-SBE-β-CD包合物的成功制备大大提高了GLA在水介质中的溶解度。
2.4 DSC分析GLA曲线在225 ℃处有一个尖锐的吸热峰,与其熔点相对应。SBE-β-CD在300 ℃出现一个不规则的峰,是降解过程的结果。与纯GLA和SBE-β-CD相比,物理混合物中GLA在225 ℃的吸热峰减弱,SBE-β-CD的吸热峰增强。GLA-SBE-β-CD的热图显示,纯GLA的原始吸热峰消失,表明GLA完全包合在SBE-β-CD中。见图2。

a:GLA-SBE-β-CD包合物; b: GLA-SBE-β-CD物理混合物; c:SBE-β-CD; d: GLA
2.5 XRD表征GLA图谱显示多个清晰的峰,表明GLA以典型的晶线形式存在。相反,SBE-β-CD表现为一条平坦的曲线,在扫描范围内只有一次微弱的上升,表明其为非晶态。在其物理混合物的衍射图中观察到了GLA的主要特征峰,但强度较低。从GLA-SBE-β-CD图谱可以看出,GLA的大部分特征结晶峰已经消失。这一现象表明,GLA不再以晶态存在,而是包含在SBE-β-CD空腔中。见图3。
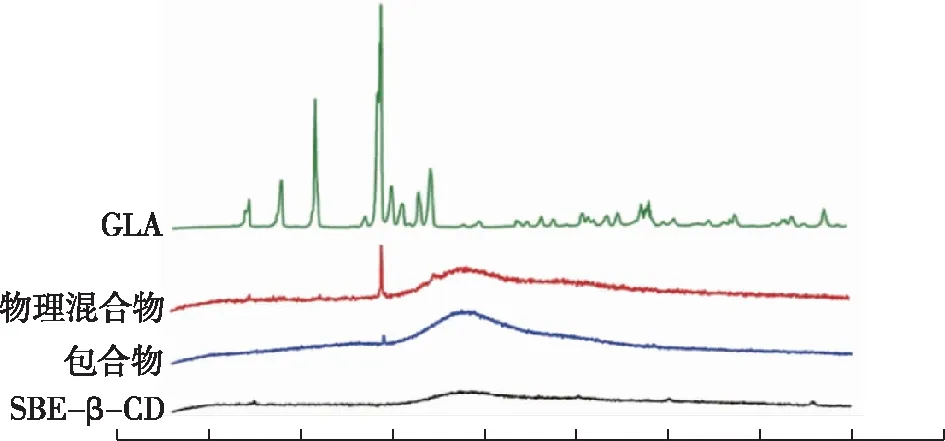
图3 不同制备方法下的混合物和蓝萼甲素A的的X-衍射图
2.6 SEM分析SBE-β-CD为边界光滑的球形,而GLA为尖角状和光滑的柱状晶体。在它们的物理混合物照片中,明显观察到SBE-β-CD的球形和GLA的柱状晶体。在GLA-SBE-β-CD的照片中,SBE-β-CD和GLA的特殊形态特征已不存在。GLA-SBE-β-CD颗粒呈无定形,略大于GLA和SBE-β-CD。GLA-SBE-β-CD的表面形貌明显不同于物理混合物、GLA和SBE-β-CD,已形成了一种新的物相。见图4。

a:GLA原料药;b:SBE-β-CD;c:GLA原料药和SBE-β-CD物理混合物;d:GLA-SBE-β-CD包合物图4 不同制备方法下的混合物和蓝萼甲素A的的扫描电镜图
2.7 人源癌细胞抑制作用研究不同浓度的GLA和GLA-SBE-β-CD对A549、Hela、HepG2、SiHA细胞的抑制率见图5。GLA及其包合物对这4种肿瘤细胞均有明显的抑制作用,GLA-SBE-β-CD对这4种肿瘤细胞的抑制率明显高于GLA。浓度为10 μg/mL GLA-SBE-β-CD组对A549和HepG2的抑制率为(84.18±4.26)%和(76.29±1.29)%,略低于对Hela(88.02±2.90)%和SiHA(90.38±4.1)%的抑制率,也说明了GLA在抑制癌细胞上具有一定的选择性,且具有浓度依赖性。

a:A549;b:Hela;c:HepG2;d:SiHA图5 蓝萼甲素A及磺丁醚-β-环糊精包合物对A549、Hela、HepG2、SiHA细胞抑制作用
3 讨 论
本研究采用超声波法制备了GLA-SBE-β-CD包合物,在制备过程中考察了GLA和SBE-β-CD的比例对包封率的影响,同时通过Box-Behnken设计法优化了制备中温度、SBE-β-CD浓度和包合时间等条件对包封率的影响,筛选出包封率最高时的制备条件,确定了最佳制备工艺,然后又通过XRD、SEM和DSC对GLA-SBE-β-CD包合物进行表征,结果表明GLA与SBE-β-CD形成包合物。最后选取4种常见的致死率较高的癌细胞,初步考察GLA-SBE-β-CD包合物对其增值的抑制效果。实验表明GLA-SBE-β-CD包合物的制备改善了GLA的水溶性,且GLA-SBE-β-CD包合物的包封率达到87%。GLA-SBE-β-CD包合物的抑制癌细胞生长效果明显高于未被包合的GLA,表明该方法制备的GLA-SBE-β-CD包合物能够有效提高GLA的溶解度,加速了癌细胞的凋亡。同时本研究选用的4种癌细胞均属于临床诊断中较为常见的癌细胞,所制备的GLA-SBE-β-CD包合物为以后GLA活性成分的临床用药提供了一定理论依据。
综上所述,本研究通过使用包合物的制剂手段使GLA活性成分在临床抗肿瘤中具备一定的应用潜力和开发价值,此外还有待深一步对其体内的药代动力学进行研究,为临床用药提供剂量参考。
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10 09:11:20
承德医学院学报(2022年2期)2022-05-23 13:01:36
中成药(2018年8期)2018-08-29 01:28:08
中成药(2018年5期)2018-06-06 03:11:49
中成药(2018年4期)2018-04-26 07:12:43
中成药(2017年12期)2018-01-19 02:06:56
中成药(2017年5期)2017-06-13 13:01:12
中国洗涤用品工业(2015年9期)2015-02-28 19:03:04
郑州大学学报(工学版)(2014年6期)2014-03-01 04:21:28
食品科学(2013年13期)2013-03-11 18:24:19
