
单原子催化剂在电催化还原CO2中的应用进展*
2021-12-14 10:39马春路孙艳芳吕宏虹刘庆龙沈伯雄
功能材料 2021年11期
马春路,孙艳芳,吕宏虹,2,张 慧,李 聘,刘庆龙,沈伯雄
(1. 河北工业大学 能源与环境工程学院,天津 300401;2. 天津市清洁能源利用与污染物控制重点实验室,天津 300401;3. 河北工业大学 化工学院,天津 300401; 4. 南开大学环境科学与工程学院,天津 300350)
0 引 言
由于大气中CO2浓度的增加和化石燃料储量的有限,迫切需要开发设计出高效技术,将温室气体CO2转化为有价值的化学物质储存起来。目前国内外学者已展开了大量的研究,开发捕获、利用和储存CO2新技术。CO2还原反应(carbon dioxide reduction reaction, CO2RR)是将CO2转化为工业化学品和燃料的一条很有前途的途径。此项技术不仅可以解决像化石燃料等不可再生资源短缺的能源危机,还可以减轻CO2对环境所造成的负担,将CO2进行资源化无害化处理。CO2RR包括化学、电催化、光催化和生物等方法,其中电催化方法具有操作简单、占地面积小等优点。
电催化还原CO2的原理是将CO2吸附在电极-电解液界面的催化剂上,在通电条件下部分碳氧键断裂、碳氢键组合生成目标产物,产物再从催化剂表面和电解液中脱附[1-9]。电催化还原CO2不仅具备上述优点,还因其反应条件温和且易于控制而成为了研究的热点[10]。但由于CO2具有一定的稳定性,在电解质水溶液中溶解度较小,所以反应发生的过电位较高[11],因此CO2电还原的可行性关键取决于开发高活性和选择性的催化剂,降低所需的过电位。目前已经研究的催化材料可分为金属单质材料、金属化合物(如金属氧化物、合金等)、有机分子、生物催化材料以及相应的复合材料等,其中研究最为普遍的为金属催化剂(特别是过渡金属催化剂)。但就目前研究的催化材料来看,普通的金属电极在电催化过程中存在效率低等问题,如何使催化剂具有高活性以及高选择性一直是个难题。因此,由张涛团队[12]制备并提出的单原子催化剂引起人们广泛关注。单原子催化剂(single-atom catalysts, SACs)是一类仅含有相互孤立的单个原子作为催化活性中心的负载型催化剂。单原子催化剂具有高活性、高选择性、高稳定性、可以重复循环使用等特点[1],但针对单原子催化剂在CO2电化学还原方面的研究目前还处于起步阶段。本文将总结单原子催化剂在电催化还原CO2的研究进展,从单原子催化剂的制备方法、表征、电化学性质以及电催化还原CO2机理与应用几方面进行总结,并在最后对单原子催化剂目前存在的问题以及未来研究趋势提出展望。
1 单原子催化剂的制备与表征
在电化学催化还原CO2中,催化剂的种类不同,电解产生的产物也不尽相同。日本学者Azuma[2]对32种金属电极在水相中电催化还原CO2进行了研究,实验结果表明金属材料Pb、Hg、Ti、In、Sn、Cd、Bi、Hg/Cu、Sn-Cd,Sn-Zn等的电解主要产物为HCOOH/HCOO;金属材料为Au、Ag、Zn,Pd,Ga,Ni-Cd等的主要还原产物为CO;而Cu及Cu的合金(如Ag、Au、Fe、Ni、Zn、Sn等)则会同时生成CO、烷烃、醇、酸等多种还原产物。
单原子催化剂则是在空间上缩小催化剂的尺寸使其达到原子级水平,从而增加催化剂与反应物的接触面积,提高催化材料的利用效率,增强催化活性,从而提高催化效率。为了防止单原子催化剂因团聚而降低催化效能,通常用载体负载单原子催化剂,负载后的单原子催化剂具有高选择性、高活性的优异催化性能等特点,目前常用的载体材料包括金属氧化物、金属单质、石墨烯等碳材料和金属有机骨架。载体不同,催化剂性能不尽相同,同样制备方法不同也会影响催化剂的性能[3]。下面我们将介绍几种典型的单原子催化剂的制备方法。
1.1 单原子催化剂的制备方法
传统负载型催化剂的制备重点是需要保证催化剂的活性组分成功负载到基底材料上,与传统负载型催化剂制备方法相比,单原子催化剂的制备不仅要保证催化剂的活性组分成功负载,还要控制催化剂活性组分的添加量,以确保制备出来的催化剂达到原子级水平。目前常用的制备单原子催化剂的方法有以下几种:
(1)共沉淀法。该方法以金属离子为前驱体,通过向前驱体水溶液中滴加沉淀剂使其沉淀来制备负载均匀的单原子催化剂。Qiao及其团队[4-5,12]通过向碳酸钠(Na2CO3)溶液中逐滴加入氯铂酸(H2PtCl6·6H2O)和硝酸铁(Fe(NO3)3·9H2O)混合溶液的方式得到单原子催化剂Pt1/FeOx(“1”代表Pt的原子个数为1)。通过研究发现,Pt负载量为0.17 %(质量分数)时(Pt/Fe原子比为1/1430),Pt1/FeOx具有较高的催化活性,催化剂表现的效果最好,而随着Pt负载量的增加,出现单原子Pt的团簇,催化剂的性能有所降低。共沉淀法是一种应用广泛且技术成熟的制备方法。该制备工艺简单,但所用的试剂量不宜控制,容易产生团聚或组成不够均匀,影响催化剂效果。
(2)逐步还原法。逐步还原法需要先制备一种金属纳米团簇(M-NCs),然后通过离子交换等反应将另一种金属原子负载到纳米团簇表面,从而制得金属纳米团簇负载的单原子催化剂。张海军[7]及其团队通过逐步还原法制备成功制备了单原子催化剂Au/Pd-NCs。首先采用醇还原法制备了聚乙烯吡咯烷酮(PVP)保护的Pd-NCs分散体,然后将四氯金酸(HAuCl4)和PVP水溶液快速加入到含L-抗坏血酸的Pd胶体分散液中制备Au/Pd-SACs。与Au-NCs、Pd-NCs、Au/Pd合金这几种催化剂相比,Au/Pd-SACs在葡萄糖氧化中表现了超高的催化活性。该方法可以较好的将金属单原子负载到纳米团簇表面,但制备过程复杂,且制成的单原子催化剂稳定性较差,难以工业化生产使用。
(3)浸渍法。该方法是将载体浸入到催化剂前驱体溶液中,通过载体表面的静电吸附使金属离子负载在载体表面,并通过蒸发、干燥及还原等工艺制备催化性能良好的单原子催化剂的方法。Melanie[8]及其团队成功将单原子Pt负载到氧化铝(Al2O3)表面,制备了Pt/Al2O3-SACs用于CO2还原。证实了在惰性载体上制备的单原子催化剂同样具有催化活性。现在经常选用金属氧化物、一维纳米线及二维纳米片等作为浸渍法制备单原子催化剂的载体。该方法操作简单,也是一种应用广泛制备单原子催化剂的方法,但金属负载量较低,当金属浸渍量大时,浸渍后金属原子在载体上容易造成分布不均匀的现象[8]。
(4)质量分离软着陆技术。通过高频激光蒸发金属前驱体,使金属汽化,利用质谱仪精确调控,使不同原子个数的金属团簇负载到载体表面。该方法使用大量选择的分子和原子束精确控制金属粒子的大小,并与超高真空表面科学(UHV)手段相结合,精确调节载体的表面结构。Abbet等[9]研究人员采用质量选择软着陆技术将Ptn(n代表Pt原子个数)团簇负载到MgO(100)薄膜上制备单原子催化剂用于乙炔环三聚制苯反应,实验中对于小尺寸团簇,在300 K左右会有苯的生成,随着n的增加,当n达到7以后,生成苯的温度会达到430 K左右时(1≤n≤30),单个Pt原子只有负载到MgO(100)上才具备催化活性。质量分离软着陆技术在制备支撑金属团簇甚至囊泡方面具有强大的优势,因为它可通过分离选择来精确控制金属物种的大小,但由于该方法对于实验条件较为苛刻,且催化剂的产率低限制其广泛应用,无法实现大规模生产。
(5)火焰喷雾热解法(FSP)。火焰喷雾热解法是制备催化剂的一种新型方法,是一种有效合成均匀大小的金属纳米颗粒的技术。制备过程是将金属盐溶液以水雾状喷至燃料气体燃烧产生的高温火焰中,溶剂蒸发与金属盐热解同时发生,溶液中两种或多种物质便负载到一起,可一步制得催化剂。丁仕鹏[13]等研究人员尝试通过火焰喷雾热解(>1000 ℃)在4种氧化物载体上合成Pt-SACs,在这4种载体(Al2O3、二氧化硅(SiO2)、二氧化钛(TiO2)和二氧化锆(ZrO2))中,ZrO2具有最强的稳定作用,与传统浸渍法制备的单原子Pt催化剂相比,火焰喷雾热解合成的催化剂在CO氧化,甲烷燃烧和甲烷部分氧化反应中表现出优异的催化性能。火焰喷雾热解法优点是在起始阶段就可达到原子水平的均匀混合;可通过控制合成条件控制催化剂的形貌和颗粒,如通过控制火焰温度来制备不同金属负载量的催化剂。因为火焰温度较高还防止金属原子的蒸发和复合,该方法过程简单、成本低、可大量生产催化剂。但由于该方法较为新颖,目前在合成单原子催化剂方面应用较少,故无法根据现有材料推断火焰喷雾热解是否确实是一种制备单原子催化剂的可行方法。
(6)原子层沉积技术(ALD)。原子层沉积法是一种可以将金属以单原子膜的形式一层一层地镀在基底表面的方法。通过简单调整原子层沉积的循环次数,就可以达到控制纳米团簇大小、形貌以及质量的目的[14]。Sun[15]及其团队通过该方法成功制备了Pt/石墨烯单原子催化剂,并通过简单地调整ALD循环数,精确地控制了Pt在石墨烯上的形貌、尺寸、密度和负载量。与传统的Pt/C催化剂相比,这些催化剂具有更高的甲醇氧化活性和更好的CO耐受性。ALD目前广泛应用与纳米材料的制备。ALD提供了精确控制催化剂尺寸跨度从单个原子,亚纳米簇到纳米粒子的能力。该种方法每次只能沉积一层原子故也称为单原子层沉积。但该方法所需的设备和材料成本较高,目前并不利于大规模生产。
(7)化学气相沉积法(CVD)。是利用气相反应,在高温、等离子或激光辅助等条件下控制反应气压、气流速率、基片材料温度等因素,从而控制纳米微粒薄膜的成核生长过程。或者通过薄膜后处理,控制非晶薄膜的晶化过程,从而获得纳米结构的薄膜材料。何宇[16]等研究人员开发了一种低温(低至450 ℃)化学气相沉积策略制备片状的开放纳米结构,其中Ni纳米粒子被Ni-N物种分散的碳层包裹(Ni-NC@Ni),在CO2RR中表现出高选择性、高电流密度等显著性能。该方法利用调节沉积的参数,可以有效地控制覆层的化学成分、形貌、晶体结构和晶粒度等,设备简单、操作维修方便,但该反应前驱体的活性较低,用量需仔细控制,常规实验中反应温度太高(850~1 100 ℃),一般载体材料承受不住。
(8)电弧放电法。该方法是通过将阳极移动到阴极来引发电弧放电,阳极在电弧产生的高温下蒸发,使得带电粒子在阴极表面沉积,从而制备所需材料。Yan[17]等研究人员采用电弧放电法在氯铂酸溶液中合成金属/碳洋葱(同圆纳米石墨壳结构)复合材料,成功制备了Pt/C催化剂。实验中Pt1@C催化剂,在温和的反应条件下,对功能化硝基芳烃的化学选择性加氢具有较高的催化活性和良好的稳定性,并且经过10次循环后依然能具有较高的催化性能。相较于其他多步合成过程,该方法一步合成,大大缩减了制备催化剂所需的时间。电弧放电法对实验条件也较为苛刻,电弧电流过低电弧不稳定,电流过高会使形成的催化剂杂质增加。而且在起弧时,温度达到了3 000~5 000 ℃,如果没有很好地让电极降温,不仅会影响到电极起弧的寿命,还很难让生成的沉淀物沉积下来。
表1总结了单原子催化剂制备方法与特点。总的来看,目前常用的制备单原子催化剂的方法主要是共沉淀法与浸渍法,由于其技术成熟、操作简单的特点,已经被广泛的研究和使用。电弧放电法具有操作过程简单、一步合成的特点,在单原子催化剂的制备中具有更加广泛的应用前景,但其制备条件的改变对单原子催化剂的催化性能产生较大影响,需要在将来的工作中进一步的研究。

表1 单原子催化剂制备方法与特点Table 1 Preparation methods and characteristics of single atom catalyst
1.2 单原子催化剂的表征
目前针对功能材料的表征技术已经被广泛研究和使用,主要包括透射电子显微镜(TEM)、扫描透射电子显微镜(STEM)、X射线能量色散谱(EDS)、电子能量损失谱(EELS)、X射线吸收精细结构谱(XAFS)、X射线衍射(XRD)、X射线光电子能谱(XPS)、傅里叶变换红外光谱(FTIR)、比表面积测试法(BET)等。在电催化还原CO2中,针对单原子催化剂,研究者首先关注的就是单原子是否成功负载到载体上,其次为了更好地了解与研究单原子催化剂的作用机理,对单原子催化剂中的原子状态、活性组分、原子结构等也要进行深入研究,本文将介绍几种典型的表征技术,并论述该种技术在单原子催化剂表征方面的应用。
1.2.1 高角环形暗场像扫描透射电镜(HAADF-STEM)
STEM是一种既有透射电子显微镜功能,又有扫描电子显微镜功能的显微镜,可以利用磁透镜将电子束聚焦到样品表面并在样品表面快速扫描,通过电子穿透样品成像。高角环形暗场像(HAADF)作为扫描透射电镜(STEM)最主要的成像模式之一,给材料、物理、生物、化学等领域的显微学研究工作带来许多新的突破。成像后图像像点的强度正比与对应原子序数的平方,可实现化学成分信息分辨[18]。根据HAADF-STEM扫描结果,可准确判断出制备的催化剂是否为单原子催化剂,是表征单原子催化剂的关键技术之一。Bi[19]等研究人员通过HAADF-STEM结合EDS揭示了Pt1/MoC中铂原子的形成(图1),像差校正HAADF-STEM图像直接显示了铂单原子,证明不仅成功将Pt负载载体上,还成功制备出了单原子催化剂。

图1 Pt1/MoC (a)HAADF-STEM图像; (b)EDS-Mo、C和Pt映射; (c)经像差校正的Pt1/MoC STEM-HAADF图像[19]Fig 1 (a) HAADF-STEM image; (b) EDS Mo, C, and Pt mappings; and (c) aberration-corrected STEM-HAADF image of Pt1/MoC[19]
1.2.2 X射线光电子能谱(XPS)
X射线光电子能谱是分析物质表面化学性质的一项技术。XPS可测量材料中元素组成、经验公式、元素化学态和电子态。光电子谱峰的能量和强度可用于定性和定量分析所有表面元素(氢元素除外)。特别是可以用于包括绝缘体,半导体等所有粉末,固体或者薄膜等材料的表面分析手段。通过该项技术我们可以判断单原子在发生催化反应时,元素的价态状态,便于对催化反应机理的研究与分析。Bi[19]等研究人员通过对单原子催化剂Pt1/MoC扫描,分析催化剂中元素的存在状态,如图2所示。Pt1/MoC催化剂Mo 3d的XPS数据表明Mo以Mo4+和Mo2+状态存在(图2(a))。图2(b)描绘的Pt 4f XPS显示原子分散的Pt处于Ptδ+状态,表明Pt1/MoC中Pt单原子的独特电子结构。XPS也是研究石墨烯中氮掺杂效应的标准技术。Wang[20]及其团队利用N-石墨烯的XPS谱中N1s光谱来确定氮的配置。N1s光谱通常可以被分解成几个单独的峰,这些峰被分配到吡啶氮(398.1~399.3 eV)、吡咯N(399.8~401.2 eV)和四元N(401.1~402.7 eV)。
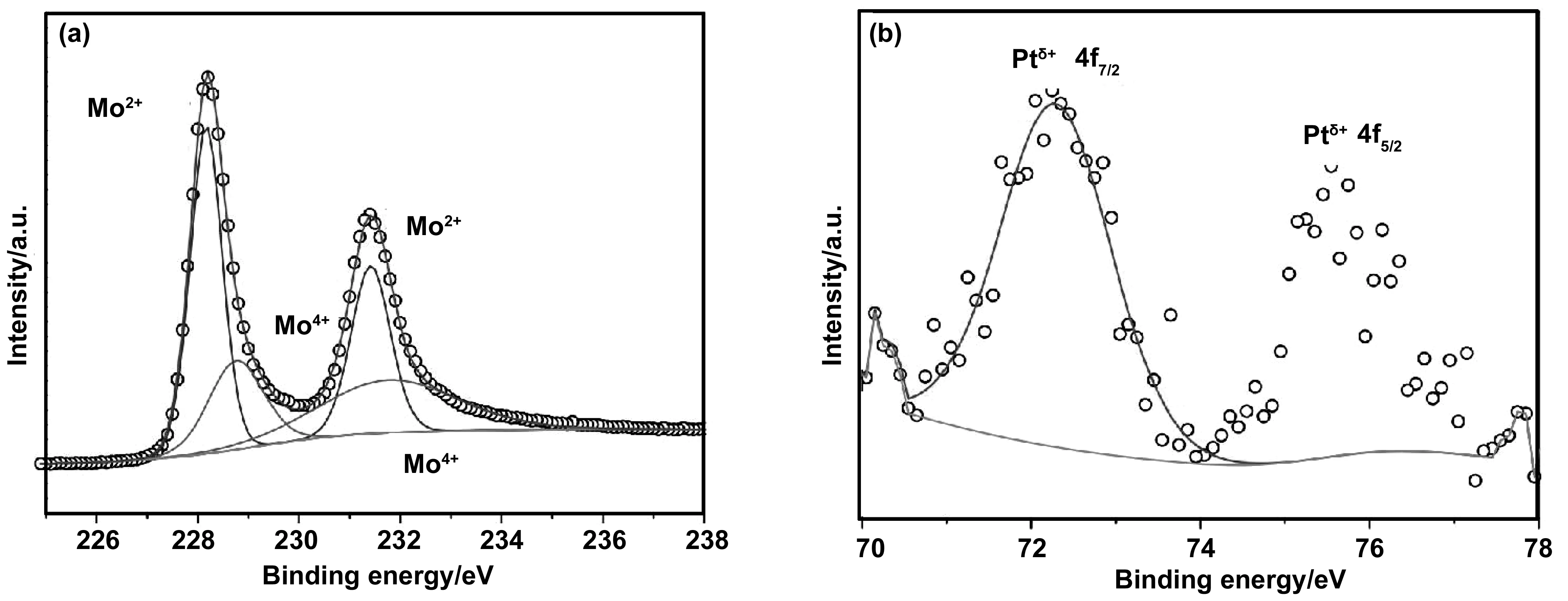
图2 Pt1/MoC的XPS(a)Mo 3d和(b)Pt 4f[19]Fig 2 XPS of Pt1/MoC: (a) Mo 3d and (b) Pt 4f [19]
1.2.3 X射线吸收精细结构谱(XAFS)
为了深入了解单原子催化剂的催化性能,不仅需要对单原子在载体表面的分布进行研究,还需要对负载的单原子进行微观结构的观察。XAFS则成为描绘局部结构最有力的工具。通常将XAFS分为近边结构谱(XANES)和扩展的X射线吸收精细结构谱(EXAFS)。XANES为吸收边前10 eV到边后50 eV的能量范围,主要用来探测吸收原子的价态、化学成键和立体配位情况。EXAFS为吸收边后50~1 000 eV能量范围内的振荡结构,主要用来探测周围原子种类、配位数、键长和无序度。
XANES表征技术可以用来确定催化剂几何和电子结构、表征d-带特性、测定配位电荷、提供包括轨道杂化、配位数和对称性等结构信息。Bi[19]等研究人员在Pt/MoC的X射线吸收近边结构(XANES)光谱中观察到,发现从Pt箔到Pt1/MoC发生的可见蓝移,表明Pt1/MoC中的Pt单原子此时具有正电荷。而后又采用原位漫反射红外傅里叶变换光谱(DRIFTS)检测Pt/MoC上的CO化学吸附,发现随着Pt尺寸的减小,CO吸附发生明显的偏移。进一步证实了Pt1/MoC催化剂中Pt原子的精细分离。
EXAFS不会受其他元素的干扰,对不同的元素的原子,可由吸收边位置不同,而得以分别研究。孙淑辉[15]及其研究团队采用XANES研究通过ALD技术制备的催化剂样品进行表征,图3中展示出了一系列Pt试样在PtL3边缘附近边缘结构(XANES)的X射线吸收。对比样品包括3个不同Pt沉积周期的ALD样品、铂金属箔和Pt/C商业催化剂。在Pt-ALD样品中观察到的XANES模式在强度上增长,并且随着循环次数的增加而变得更尖锐,这表明晶体的顺序增加并接近Pt箔。图3(b)绘制了各种情况下EXAFS区域的傅里叶变换。结合图表发现50ALDPt/GNS样品具有很强的Pt-C或Pt-O贡献,Pt-Pt键距离与完美的Pt金属有很大的偏差,表明在50ALDPt/GNS样品中,只有非常小的Pt团簇和单个原子占主导地位。较小的Pt团簇可以提供更多的可用催化位置,从而更加证实了Pt单原子在样品中起主要作用(例如在50ALDPt/GNS)。XAFS分析表明,Pt原子5d轨道状态的低配位和部分未占据密度是性能优异的原因。
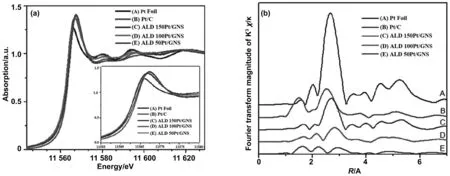
图3 (a)PtL3边缘的归一化XANES光谱和(b)样品EXAFS的K3加权傅里叶变换光谱[15]Fig 3 X-ray absorption studies: (a) the normalized XANES spectra at PtL3 edge and (b) the K3-weighted Fourier transform spectra from EXAFS of samples[15]
1.2.4 电化学性质测量
为了更好的了解CO2还原反应动力学,一些研究学者还对催化剂进行了Tafel图(塔菲尔图)和电化学阻抗谱(EIS)的研究。
Tafel图反映了电流密度的对数与过电势之间的关系(超电势(η)与电流密度(i)有如下关系:η=a+b*log|i|,a、b称为塔菲尔常数),表示为了达到一定的电流需要改变电极电势的程度。Pan[21]及其研究团队对NFe-CNT/CNS和NFe@CNT进行了Tafel分析(图4(a))。对于NFe-CNT/CNS,在0.19~0.39 V的过电位下,其Tafel斜率为89 mV/dec,相较于NFe@CNT的Tafel斜率较小,因此表明在CO2RR中,NFe-CNT/CNS上的CO2分子具有更快的初始电子转移能量,表示较高的动力学速率。为了进一步表示NFe-CNT/CNS在CO2RR中的优越性。Pan等[21]还进行了电化学阻抗谱(EIS)测试,如图4(b)奈奎斯特图(Nyquist图)所示,由两个半圆组成,其中在实轴上的截距归因于由溶液和电极接触电阻导出的串联电阻。第一个半圆通常表示电荷转移电阻,与从催化剂表面转移到反应物的电子数以及中间产物的形成有关;第二个半圆表示离子的能斯特扩散阻抗,与质量传递有关。NFe-CNT/CNS比NFe@CNT具有更低的界面电荷转移电阻和更低的能斯特扩散阻抗,因此电荷转移加快,质量传递加快,从而最终提高了CO2还原为CO的活性。
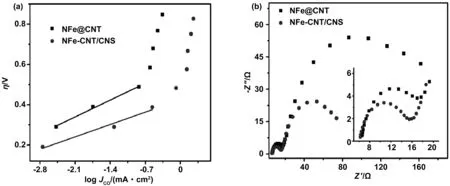
图4 在二氧化碳饱和的0.1 mol/L碳酸氢钾水溶液中(a)CO局部电流密度的Tafel图;(b)NFe CNT和NFe CNT/CNS的Nyquist图(插图示出高频区域中的放大图)[21]Fig 4 (a) Tafel plots of CO partial current density and (b) Nyquist plots for NFe CNT and NFe-CNT/CNS in CO2-saturated 0.1 mol/L KHCO3 aqueous solution[21]
2 单原子催化剂电催化还原CO2的影响因素与研究现状
电化学中的CO2还原CO2RR是一个热力学上升过程,是通过使CO2失去2、4、6、8电子等多电子来完成,涉及几个中间体间的复杂多电子转移过程,需要高活性的电催化剂来加速反应。同时,反应过程中的析氢反应(HER)相互竞争,使得还原过程更加复杂。而不同种类的催化剂下生成的产物也不尽相同,由简单的一氧化碳分子到长链的碳氢化合物、醇、酯等,其中最为常见的还原产物有一氧化碳、甲酸、草酸、甲醛、甲醇、甲烷、乙烷、乙醇等[22](反应方程式见表2[23])。因此,合成具有高选择性的理想催化剂至关重要。
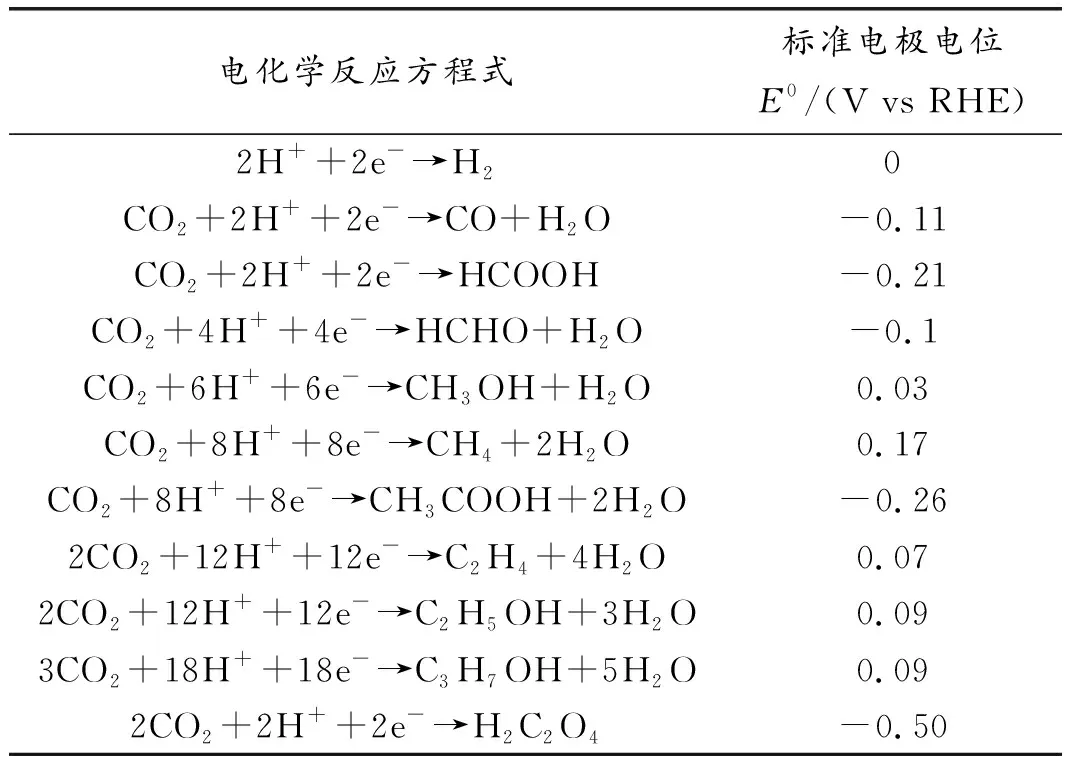
表2 电化学反应方程式Table 2 Electrochemical reaction equation
2.1 单原子催化剂电催化还原CO2的影响因素
近30年的在金属化合物和无金属异原子掺杂的碳材料作为CO2RR的电催化剂方面的研究取得重要进展。在碳载体上的单原子催化剂由于其提高了原子利用率、具有不饱和金属配位和良好的传导率作为优秀的化学反应电催化剂成为了研究热点。 单个原子可以分散在不同碳材料上,如石墨烯、碳纳米管(CNT)、石墨氮化碳(g-C3N4)和共价有机骨架材料(COFs),对于原子分散的金属中心,它们的反应中间体的吸附和解吸行为高度依赖于周围的配位环境和来自碳基质的长距离电子插层,碳材料作为理想的基底,具有较高的活性和选择性。综合研究表明除催化剂的选择性和稳定性外,金属原子周围的配位数、载体的比表面积和缺陷也是影响单原子催化剂电催化还原CO2的重要因素(表3)。目前针对单原子的催化剂的研究,也主要集中于这几个方面,接下来将主要从这几方面展开论述单原子催化剂电催化还原CO2的效能和应用。
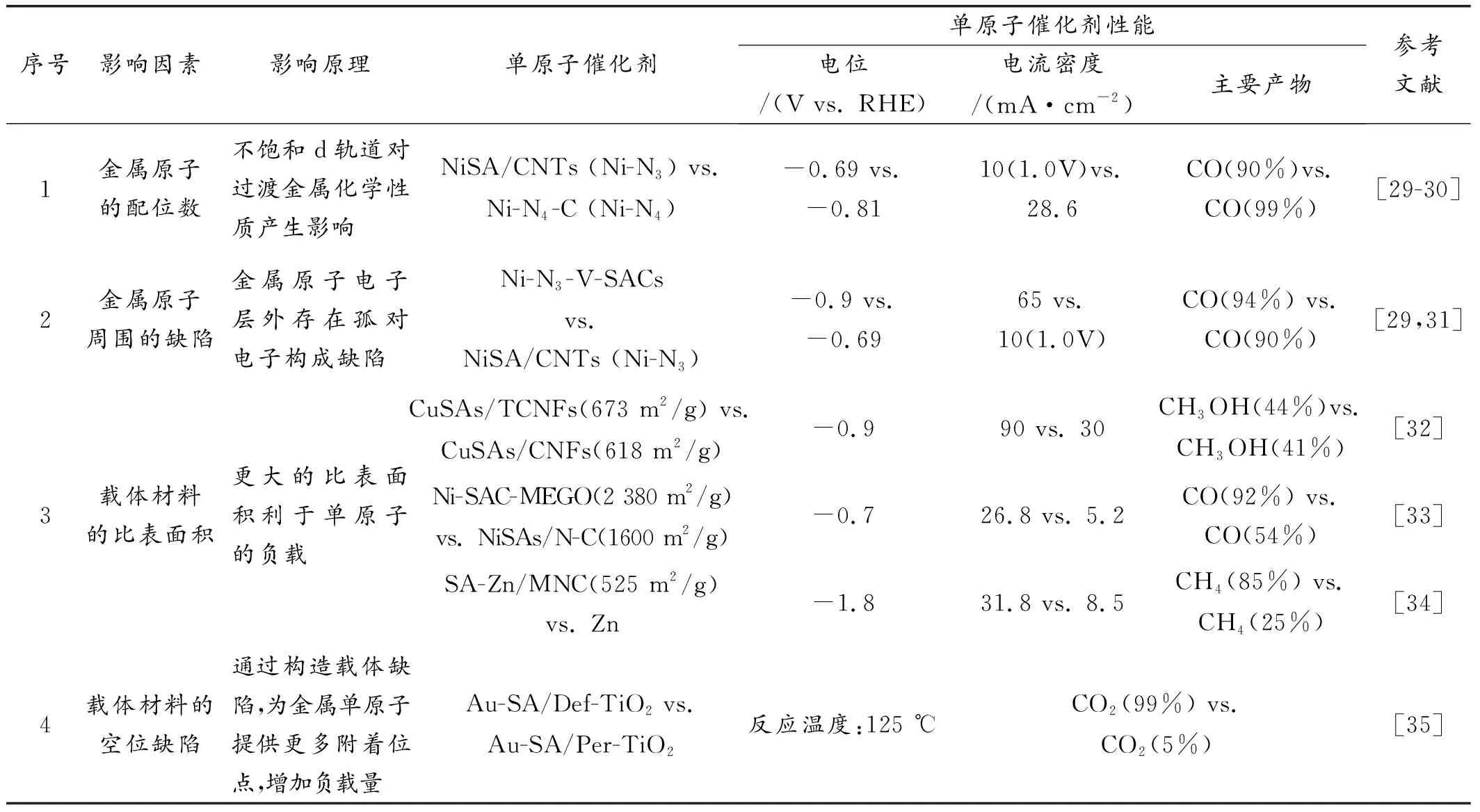
表3 影响单原子催化剂催化的因素Table 3 Factors affecting SACs catalysis
2.1.1 单原子催化剂的高选择性和稳定性
过渡金属(M)和氮原子(N)共掺炭(C)形成的催化剂M-N-C,特别是多种氮掺杂碳负载过渡金属催化剂(M-N-C)也已被证明是CO2RR的良好催化材料[24-25],这是由于金属原子与氮配位形成了M-N配合物,相比于单个金属原子可增加催化剂的催化活性。Huang等[26]利用单原子Mo负载到超薄N掺杂石墨烯上制成单原子催化剂Mo@NG。与NG相比,在将CO2电催化还原为甲酸(HCOOH)中表现了超高的电催化活性。在研究中发现,随着电位的升高,在NG和Mo@NG作用下产生的HCOOH的FE差距相当明显,在Mo@NG中的FEHCOOH在每个电位下几乎都是NG中FEHCOOH的两倍,而在-1.4 V时更是接近3倍。这是因为NG为Mo提供了适当的锚固点,使得Mo@NG具有较宽的负电位区,比传统的N掺杂石墨烯产量更高,具有更好的催化活性。
Cheng等[27]通过多步热解的方法将Ni镶嵌到氮掺杂的碳纳米管上,成功合成了单原子催化剂NiSAC@N-CNTs[28],与金属纳米颗粒相比(Ni@N-CNTs),NiSAC@N-CNTs表现出更明显的CO2还原效率,证明了其对CO2还原具有极高的活性和选择性。在-0.7 V时催化剂的电流密度为23.5 mA/cm2,此时CO的FE可达到91.3%,NiSAC@N-CNTs的高催化活性和稳定性是由于N原子被锁定的活性Ni-N配位引起的,即使在电解12 h后,NiSAC@N-CNTs对CO2的还原表现出显著的稳定性。
2.1.2 金属原子周围的配位数
由于过渡金属的活性主要来源于它们的不饱和d轨道,合理选择活性金属中心是调节CO2RR活性和选择性的最直接途径。金属原子周围的配位数对催化剂的催化性能会产生显著影响。Fan等[29]通过激活碳纳米管中的Ni粒子,在碳纳米管表面形成了用于电催化还原CO2的Ni单原子催化剂(NiSA/CNTs)。与N掺杂碳纳米管(NC)相比,NiSA/CNTs在CO2饱和的KHCO3溶液中表现出更高的电催化电流和良好的选择性。NiSA/CNTs的COFE在0.69 V过电位下为90%,在1.0 V(相对RHE)下,分电流密度为10 mA·m2。NiSA/CNTs具有较好催化活性的原因是在其活性部分中存在的低配位NiN3吡咯位,使得催化剂在0.8 V(vs.RHE)下连续电解10 h后,仍然具有显著的稳定性。Li等研究人员[30]对Ni-N4-C与其他3种样品(N-C、Ni@N-C、Ni@N-C-Glu)进行比较,DFT计算表明,COOH*在NiN4上的吸附在热力学上比H*在高过电位下更有利,从而导致CO2RR的高FE。随着电压的增加Ni-N4-C表现了最高的电流密度,展示了良好的导电性,且随着电压的增加CO的法拉第效率(FE)也在增加,在-0.81 V时,电流密度为28.6 mA/cm2,COFE达到最大99%。
Guo[36]及其团队通过限制不同电催化反应之间的电位差,描述了氮掺杂碳基上8种金属单原子不同配位数对CO2RR的选择性,如下图5所示,图中总结了M-Nx位点上的UL(CO2RR)-UL(HER)。二者的差值用来描述该种配位的金属对反应CO2RR与HER的选择性,较大的UL(CO2RR)-UL(HER)对应于较高的CO2RR选择性。如图所示具有相同金属中心的M-N4位点与其自身的M-N1,2,3相比,UL(CO2RR)-UL(HER)的差值更大,所以对CO2RR具有更高的选择性。而在这几种金属中,CO2RR在Fe-、Ni-、Cu-、Pd-和Pt-N4上与HER相比具有更强的竞争力。Ni和Pt两种金属在实验中受析氢反应的影响较小,Fe-N4和Ni-N4在生成CO实验中表现出了最高的催化性能。
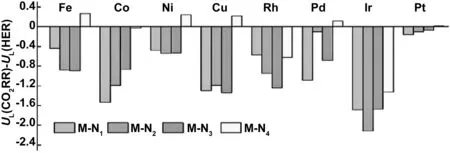
图5 限制CO2RR与HER的电位差(UL(CO2RR)-UL(HER)>0对应于较高的CO2RR选择性)[36]Fig 5 Limiting potential difference between CO2RR and HER. UL(CO2RR)-UL(HER)>0 corresponds to high CO2RR selectivity[36]
2.1.3 金属原子周围缺陷
除了金属原子在单原子催化剂中的配位环境外,M-N物种周围的缺陷对单原子催化剂的性能也会产生极大的影响。Rong[31]等研究人员通过改变M-N周围缺陷,如通过构造空位缺陷来提高催化剂的活性。与Ni-N4和NC相比,Ni-N3-V-SACs中的空位缺陷可以显著提高CO的电催化活性,在-0.9 V时,CO的FE超过90%,达到65 mA/cm2的高电流密度。
2.1.4 载体比表面积
碳材料由于其原料来源广泛、结构可控、化学稳定性好、导电性好而且具有高比表面积等优点[23],成为了更为普遍的催化剂载体。碳基材料的性质对催化剂的效率也会产生影响,若碳基材料具有较高的比表面积,就会为金属原子提供大量的锚定位点,增加金属原子的负载量,增强催化剂活性。研究学者[30]通过电纺聚丙烯腈膜和Cu离子的热解,以ZIF-8为多孔剂,在通孔碳纳米纤维上合成了原子分散的CuN4(CuSAs/TCNFs)。在CuSAs作用下甲醇和CO几乎是唯一的液相和气相产物,它产生甲醇的FE可以达到44%和50h以上的长期稳定性的甲醇。与碳纳米纤维相比(CNFs),TCNFs引入通孔结构,增加了材料的比表面积,使得活性中心的位点增加,所以与CuSAs/CNFs相比,CuSAs/TCNFs电流密度随着电压的增加显著增强,特别是在-0.8 V以后,其电流密度是CuSAs/CNFs电流密度的两倍不止。Cheng[33]及其团队在微波剥离氧化石墨烯上开发了Ni-SAC(Ni-N-MEGO),成功地定制了具有Ni单原子的多孔碳的边缘结构,多孔结构的高比表面积为Ni原子的负载提供了大量的锚定位点,实现了约6.9 %(质量分数)的单原子负载,在0.59 V的过电位下表现出53.6 mA/mg的质量活性和92.1%的高选择性。DFT结果表明,高的CO2RR活性来源于边缘上不饱和Ni单原子的高负载。Karapinar研究团队[30]在ZIF-8衍生的微孔N掺杂碳上合成ZnN4(SA-Zn/MNC),与普通的Zn电极相比,在生成CH4上具有更高的催化活性,在-1.8 V时,SA-Zn/MNC上部分电流密度达到31.8 mA/cm2,产生的CH4的FE最大值达到了85%,且催化剂在测试35 h后仍然具有一定相当的稳定性。
2.1.5 载体材料的空位缺陷
构造载体的空位缺陷也会影响催化剂的催化性能。为了避免金属单原子在碳基表面重新团聚,可以在载体材料上构造缺陷[37-38]。缺陷的引入可以改变载体材料的物理和化学性质,影响材料表面活性位点的密度和活性,从而提高碳基催化剂的整体电催化性能。Wang[35]及其团队通过对TiO2载体表面进行氧空位缺陷修饰,获得了富含缺陷态的载体材料,并利用该载体材料制备了Au负载的单原子催化剂(SA-Au/TiO2),提出了一个三中心Ti-Au-Ti的稳定结构,它不仅可以支持孤立的单原子Au的稳定性,还可以通过降低能量势垒和消除孤立金原子上的竞争吸附来提高催化剂的性能。
2.2 单原子催化剂电催化还原CO2的研究现状
目前单原子催化剂电催化还原CO2的研究处于起步阶段,还有许多问题需要克服和研究。由于单个原子的表面能量和反应性很高,导致单原子材料聚集,从而降低反应效能。因此,目前关于CO2电化学还原研究热点是通过碳支持单原子材料从而提高催化性能,包括M-N-C催化剂、异质分子催化剂和COFs。M-N-C具有短程有序长程无序结构,使其在CO2RR中具有相对理想的理论活性和对电解产物的选择性,但由于缺乏精确可控的合成方法,M-N-C的催化选择性的转换仍然难以测定,实现M-N-C材料的合理设计是一个重大的挑战。异质分子催化剂是金属中心和大环螯合配体组成的金属配合物,具有明确的活动中心,碳基质独特的插层在调节金属中心上中间体的吸附能方面起着重要作用,在某些情况下,催化剂中特定的大环配体和基质在实现CO2RR中都是必不可少的。与通常包含复杂配位环境的M-N-C催化剂不同,异质分子催化剂由于其均匀和明确的结构而具有明显的优势,促进了在分子水平上对CO2RR机制的理解。COFs是一种基于可逆共价键的交联刚性芳香族分子的多孔材料,具有扩展的π共轭结构、可调组成和孔隙度,继承了小分子的活性和选择性,弥补了异质和均匀电催化剂之间的差距。同时由于π共轭结构和发达的孔隙度,提高了电导率和传质效率,与高温合成的M-N-C催化剂相比,COFs的明确结构为识别活性位点和相应的催化机制提供了更精确的信息,但是分子催化剂由于制备工艺过程比较复杂。目前研究的异质分子催化剂中,以碳材料为基底材料的单原子催化剂催化效能最为理想,这是由于碳材料具有成本低、来源广、对环境友好、稳定性好等优点。而杂原子掺杂的碳材料与高活性的金属材料进行组合形成的单原子催化剂可以充分发挥各成分的优势,碳材料可以提供大的比表面积,有利于金属原子的负载,提高金属原子的利用率。杂原子尤其是N金属的配位作用与金属原子形成稳定的催化剂,从而有利于提高CO2的转化效率[23]。根据产物分子中的碳原子数可以将产物分为C1产物(包括一氧化碳、甲酸、甲烷等)和C2产物(如乙烯、乙醇等)和C3产物(如丙醇等),其中一氧化碳和甲酸最为实用。借助单原子催化剂独特的几何和电子特性, 可以显著增强CO2还原过程活性中间体(如*COOH、*OCHO 等)的结合能, 进而促进CO2的选择性还原,同时有效抑制了析氢副反应的发生[39]。因此,结合不同维度的多孔碳材料,采用不同方法设计性能优异的催化剂以降低过电势, 提高反应选择性和稳定性是单原子电催化还原CO2研究的重点,单原子催化剂在高效电催化还原CO2反应中展现出巨大潜能。
3 结 语
近些年来,随着温室效应和能源枯竭等环境问题的显现,使得电催化还原CO2成为众多研究学者的研究热点。因为该项技术不仅可以减少空气中的CO2,减轻全球气候变暖带来的环境负担,电解的多种产物还可以通过集中收集作为能源,将过剩的电能转化为化学能储存起来,缓解能源短缺带来的危机[40]。
与普通催化剂相比,单原子催化剂具有高活性、高选择性的优点,可大幅度提升催化剂的催化性能,拥有广泛的应用前景。虽然目前关于单原子催化剂的研究报道已有很多,但仍存在以下问题需要进一步研究:
(1)单原子催化剂上的金属单原子负载量问题。虽然实现了百分百的利用效率,但由于已达到原子水平,金属原子的表面能等相应提升,增加了单原子的不稳定性,无法准确控制其负载量,使得实际的实验效果与理想的理论计算还是有一定的差距;
(2)目前研究仍然是处于实验室的研究阶段,由于成本及技术要求等问题,还无法应用于大规模的工业生产,将来更多的应用研究需要进一步加强;
(3)负载基底材料的选择。基底材料的性质在一定程度上影响单原子的催化性能,比如,当基底材料富含含氧官能团时,会将单原子氧化,降低单原子的催化效率,造成CO2RR的效率低、选择性差、过电位高等[40]。
综上,在今后研究中应充分发挥单原子催化剂的优点,继续研发更加成熟稳定的合成技术,增加单原子催化剂的选择性、稳定性;构造结构缺陷载体材料,避免单原子在载体材料上的迁移和团聚现象,降低催化效率;深入对多相催化剂机理的研究,通过改变条件等方式减少副作用的影响;增加非贵重金属在单原子领域的研究,减少材料成本,增加大规模工业生产的可能。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
第一财经(2019年8期)2019-08-26
中国有色金属学报(2018年2期)2018-03-26
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国资源综合利用(2016年7期)2016-02-03
哈尔滨医药(2015年2期)2015-12-01
学习月刊(2015年14期)2015-07-09
物理化学学报(2015年5期)2015-02-28
