
γ-Fe2 O3表面HCl 对汞的吸附和氧化机理研究
2021-12-14 08:42:20周文波牛胜利韩奎华王永征路春美
燃料化学学报 2021年11期
周文波,牛胜利,*,王 俊,李 颖,韩奎华,王永征,路春美,朱 英
(1.山东大学 能源与动力工程学院,山东 济南 250061;2.齐鲁工业大学(山东省科学院)新材料研究所,山东 济南 250014)
汞是一种常见的污染物,具有剧毒、高挥发性和化学稳定性,对人类和动物有严重危害,如神经系统疾病、皮肤病等[1,2]。煤在燃烧过程中释放出较高水平的汞,燃煤发电厂被认为是汞排放的主要人为来源[3]。2017 年8 月,《关于汞的水俣公约》正式生效,以减少人为汞的排放[4,5]。燃煤烟气中的汞主要以单质汞Hg0、氧化汞Hg2+和颗粒结合汞Hgp三种形式存在[6,7]。Hgp和Hg2+分别可以通过颗粒物控制装置和湿法烟气脱硫系统捕获[8]。然而,由于Hg0在水中的高挥发性和低溶解度,不易捕获,成为烟气中排放的主要汞形态[9],也是燃煤电厂减少汞的最大挑战。通过催化氧化Hg0为Hg2+,然后结合湿法烟气脱硫系统,被认为是减少汞排放方便又经济的选择[10]。
已有的研究表明,金属氧化物具有较高的除汞性能[11−13]。其中,磁赤铁矿等磁性材料表现出了良好的汞吸附和氧化的催化活性[14−16]。Galbreath等[17]发现γ-Fe2O3能够有效促进Hg0的氧化。Yang等[18,19]制备了Mn 改性的Mn-Fe 磁性尖晶石,具有优异的汞捕获能力。同时,HCl 被认为是燃煤烟气中的主要含氯物质,且HCl 对汞的氧化至关重要[20,21]。Liu 等[22]发 现,γ-Fe2O3上HCl 和Hg0的非均相反应促进了汞的氧化,提高了Hg0的脱除效率。但Hg0脱除过程中通过实验测定反应中间体和过渡态非常困难,密度泛函理论已越来越多地应用于元素汞在固体表面吸附和氧化机理的研究。陈佳敏等[23]模拟了Mo 掺杂Fe3O4(111)对不同汞形态的吸附,发现HgCl 和HgCl2为化学吸附,而Hg0为物理吸附。Wang 等[24]研究了Co3O4(110)表面上HCl 对汞的催化氧化机理,发现其遵循Langmuir-Hinshelwood 机制,化学吸附的Hg0与表面活性Cl 原子相互作用生成HgCl2。Zhang 等[25]研究了CeO2催化剂上HCl 多相氧化汞的反应机理,认为在Hg0→HgCl 的第一步过程中,汞的氧化遵循Eley-Rideal 机理,而在HgCl→HgCl2的第二步过程中,汞的氧化遵循L-H 机理。然而,使用密度泛函理论研究磁赤铁矿上HCl 与汞非均相反应机理的报道相对较少。
本研究采用密度泛函理论研究了HCl 对磁赤铁矿非均相催化氧化Hg0的作用机理,重点探究了Hg0、HgCl 和HgCl2等不同汞物质以及HCl 在磁赤铁矿表面的结合机理,考察了HCl 对磁赤铁矿表面吸附汞的影响机制,并对反应的中间体、过渡态和最终产物进行了分析。本研究丰富了磁赤铁矿催化氧化脱除汞的理论基础,为合理设计相应的催化剂提供了理论指导。
1 计算模型和方法
γ-Fe2O3为反尖晶石型结构,晶胞结构如图1所示,其中,晶胞参数为a=b=c=8.405 Å,α=β=γ=90°[26]。本研究选择催化氧化性强、体积增长指数低的γ-Fe2O3(001)面作为反应面[27]。此外,选择P(2×2)超胞和7 层γ-Fe2O3(001)表面原子进行计算[28],如图2 所示。计算时固定底部四层原子并使表面三个原子层弛豫。为了防止两个周期性平板之间的相互作用,使用了15 Å的真空层。
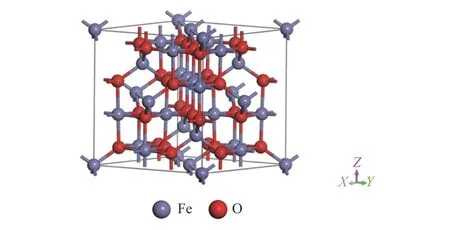
图1 γ-Fe2 O3的晶胞Figure 1 Conventional cell of γ-Fe2 O3

图2 γ-Fe2 O3表面结构Figure 2 Structure of the γ-Fe2 O3 surface
本研究计算利用CASTEP 完成,并采用广义梯度近似和Perdew-Burke-Ernzerhof (GGA-PBE) 交换相关泛函[29]。离子核采用超软赝势描述,电子波函数在平面波基组中展开,截止能量为400 eV。布里渊区K 点采用2×2×1,并考虑自旋极化。结构优化计算精度为Energy=2.0×10−5eV/atom,Max force=0.05 eV/Å和Max displacement=0.002 Å,SCF收敛标准为2.0×10−6eV/atom。通过LST-QST 的方法搜索过渡态搜索(TS),并计算振动频率验证过渡态,且TS 标准为RMS convergence:0.25 eV/Å。在10 Å×10 Å×10 Å晶格中对Hg0、HCl、HgCl 和HgCl2进行计算,优化结果见表1。计算后γ-Fe2O3晶胞参数8.357 Å,与实验基本一致[26]。
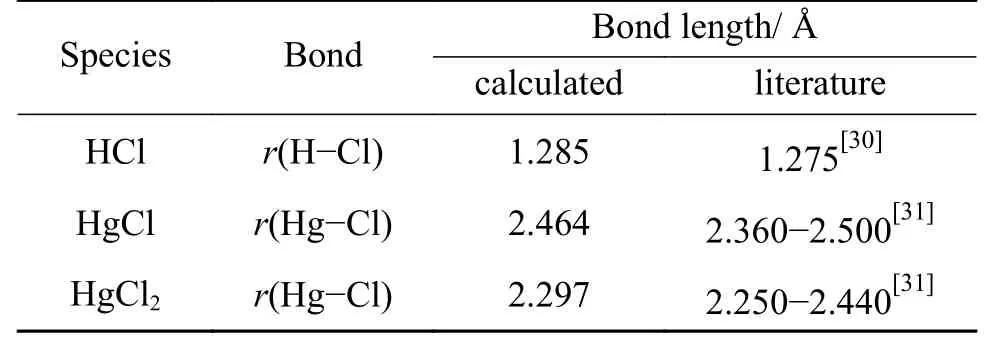
表1 键长的计算和文献值Table 1 Bond length of calculated values and literature values
表面吸附能Eads定义为吸附前后各物质总能量的变化,吸附能越大表示吸附越稳定。吸附能(ads)计算见式(1):
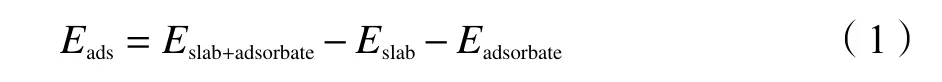
式中,Eads、Eslab+adsorbate、Eslab和Eadsorbate分别代表吸附能、底物加吸附物的总能、底物能量和吸附物能量,单位kJ/mol。
底物与吸附物间的电荷转移(ΔQ,e)定义为式(2):

式中,Qafter和Qbefore分别代表吸附物在吸附之后和吸附之前的电荷,单位e。
活化能垒(Ebarrier,kJ/mol)定义式(3):

式中,ETS和EIM分别为过渡态和中间体的能量,单位kJ/mol。
2 结果与讨论
为了研究HCl 对汞的吸附和氧化作用,计算了不同汞物种(Hg0、HgCl 和HgCl2)和HCl 的吸附性能,并通过比较Hg0在γ-Fe2O3(001)表面和预吸附HCl 的γ-Fe2O3(001)表面上的吸附能从而确定HCl 对γ-Fe2O3的汞捕获能力的影响,同时,基于能垒和反应热分析了γ-Fe2O3表面的汞的吸附和氧化过程。
2.1 Hg0 和HCl 在γ-Fe2 O3(001)表面的吸附
考虑了γ-Fe2O3(001)表面各个吸附位点,其中,吸附Hg0的最优化结构如图3 所示,相应的吸附能、结构参数和Mulliken 电荷如表2 所示,RHg-X代表Hg0与X=Fe、O 之间的距离。Hg0在γ-Fe2O3(001)表面吸附强度的增加趋势为1B <1C <1A,吸附能的变化范围为(−24.47)−(−39.26) kJ/mol,其中,最稳定的吸附构型是1A,Hg0与γ-Fe2O3(001)表面的Feoct原子键合并形成Hg−Fe 键,键长为2.919 Å。1A 的结合能为−39.26 kJ/mol,电荷转移0.14 e,表明为弱化学吸附。在1B 中,Hg0以−24.47 kJ/mol 较小的结合能吸附在γ-Fe2O3表面的O 原子位上。在1C 中,Hg0弱吸附在催化剂表面hollow 位。因此,与表面O 原子和hollow 位相比,Hg0更倾向于与γ-Fe2O3表面的Feoct原子键合,且吸附过程为放热过程,这与Guo 等[32]的研究结果一致。Mulliken电荷分析能够反映吸附过程中电荷转移情况[33]。Hg0在γ-Fe2O3表面的吸附过程,有0.04−0.14 e 的电荷转移。Mulliken 电荷都为正数,表明电子从Hg0转移到γ-Fe2O3表面。

图3 Hg0 在γ-Fe2 O3表面的优化吸附模型Figure 3 Optimized models of Hg0 adsorbed on γ-Fe2 O3

表2 Hg0 在γ-Fe2 O3表面吸附的优化参数Table 2 Optimized adsorption parameters of Hg0 adsorbed on γ-Fe2 O3
HCl 通常被认为是氧化Hg0的重要烟道气成分[20,21],且HCl 可能吸附在γ-Fe2O3表面,进而影响γ-Fe2O3吸附剂的除汞效果。因此,有必要研究HCl 在γ-Fe2O3表面的吸附,以了解整个非均相催化氧化汞的反应过程。图4 为HCl 在γ-Fe2O3表面上的优化吸附结构。HCl 在γ-Fe2O3表面吸附时,H−Cl 键发生解离,吸附后Cl−Fe 和H−O 键长度以及吸附能和电荷转移如表3 所示。在2A 中,HCl 解离吸附在γ-Fe2O3表面,并分裂成H 原子和Cl 原子。生成的H 原子吸附在O 原子上,形成表面羟基。Cl 原子与表面Fe 原子发生强烈的相互作用,形成键长为2.192 Å的Fe−Cl 键。该构型的吸附能和电荷转移分别为−73.69 kJ/mol 和0.07 e。HCl 在γ-Fe2O3(001)表面最稳定的吸附构型为2B,吸附能为−106.66 kJ/mol,反应为放热过程。在2B中,HCl 同样解离吸附在γ-Fe2O3(001)表面。Cl 原子和H 原子分别吸附在表面Fe 原子和O 原子上。H−O 和Fe−Cl 键的 键长分别 为0.983 和2.169 Å。从以上分析可知,HCl 在γ-Fe2O3表面主要是解离吸附。
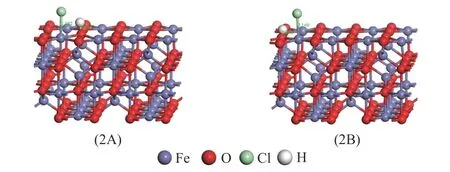
图4 HCl 在γ-Fe2 O3表面的优化吸附模型Figure 4 Optimized models of HCl adsorbed on γ-Fe2 O3

表3 HCl 在γ-Fe2 O3表面吸附的优化参数Table 3 Optimized adsorption parameters of HCl adsorbed on γ-Fe2 O3
通过考察氯化γ-Fe2O3(001)表面对Hg0的吸附,进一步探究HCl 对γ-Fe2O3脱除Hg0的影响。氯化γ-Fe2O3(001)表面吸附Hg0的稳定构型如图5所示,吸附能为−42.50 kJ/mol。汞原子被化学吸附在Cl 原子的邻近表面位置上,与表面铁原子之间的距离为2.908 Å。Hg0在氯化γ-Fe2O3(001)表面的吸附能比在纯γ-Fe2O3表面上的吸附能增加,表明HCl 促进了Hg0在γ-Fe2O3(001)面的吸附。
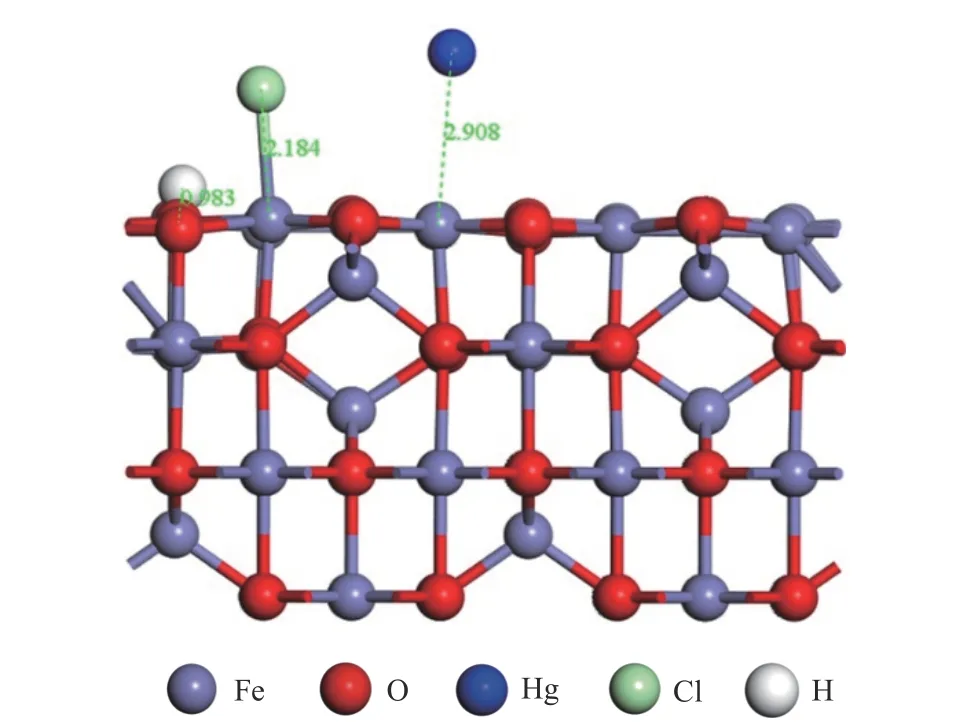
图5 Hg0 在氯化γ-Fe2 O3表面的优化吸附模型Figure 5 Optimized models of Hg0 adsorbed on chlorinated γ-Fe2 O3 surface
2.2 HgCl 在γ-Fe2 O3(001)表面的吸附
已有的研究表明,HgCl 可能是催化剂上Hg0氧化反应的中间产物[24,25]。为进一步了解Hg0催化氧化反应,本文研究了HgCl 在γ-Fe2O3(001)表面的吸附机理。考虑了所有可能的活性位点和结合方向。图6 显示了稳定的HgCl 吸附结构,相应的吸附结果见表4。HgCl 在γ-Fe2O3表面的吸附强度依次为3D >3C >3B >3A。对于最稳定的结合结构3D,HgCl 以解离结合的方式吸附,且吸附过程为放热反应,释放能量180.67 kJ/mol、电荷转移−0.12 e。3B 和3C 构型的HgCl 同样是以解离方式化学吸附,相应的吸附能分别为−141.25 和−174.54 kJ/mol。在3A 结构中,HgCl 分子以Hg 端吸附在Feoct上,Hg−Fe 键长2.545 Å。相应的吸附能 为−136.92 kJ/mol,电荷 从HgCl 分 子转移到催化剂上0.03 e,表明HgCl 也可以分子方式化学吸附在γ-Fe2O3催化剂上。上述结果表明,HgCl 在γ-Fe2O3催化剂上可以分子形式吸附和解离形式吸附。
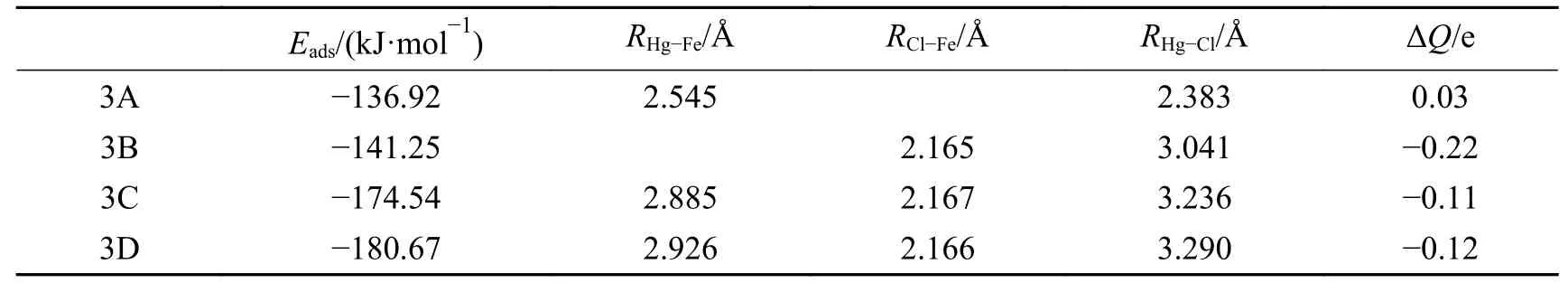
表4 HgCl 在γ-Fe2 O3表面吸附的优化参数Table 4 Optimized adsorption parameters of HgCl adsorbed on γ-Fe2 O3
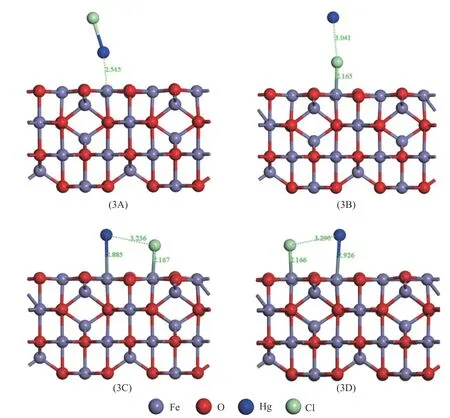
图6 HgCl 在γ-Fe2 O3表面的优化吸附模型Figure 6 Optimized models of HgCl adsorbed on γ-Fe2 O3
2.3 HgCl2在γ-Fe2 O3(001)表面的吸附
HgCl2通常被认为是Hg0与HCl 反应的主要产物。因此,有必要研究HgCl2在γ-Fe2O3表面的吸附。考虑了HgCl2水平和竖直两种吸附方式,经过结构优化,得到了三种不同的优化构型,如图7所示,相应的吸附结果见表5。HgCl2吸附的最稳定构型为4A,相应的结合能为−63.28 kJ/mol,电荷转移为0.03 e,表明为化学吸附。在4A 中,HgCl2以分子形式平行吸附在γ-Fe2O3上,其Hg 原子与表面Feoct原子成键,键长2.956 Å。在4B 结构中,HgCl2在吸附过程中发生Hg−Cl 键断裂,分裂为两个Cl 原子和一个Hg 原子。生成的Cl 原子和Hg原子与催化剂表面的Fe 结合形成Cl−Fe 和Hg−Fe键,吸附过程放热58.43 kJ/mol。在4C 结构中,HgCl2竖直吸附在催化剂表面,吸附能为−27.13 kJ/mol。对比三种吸附构型,发现HgCl2在γ-Fe2O3(001)表面吸附强度的增加趋势为4C <4B <4A,吸附能的变化为(−27.13)−(−63.28) kJ/mol。HgCl2平行吸附时,以分子形式吸附相比分裂形式吸附能更大,结构更加稳定,而垂直吸附时与γ-Fe2O3表面之间的相互作用最弱。由以上结果可知,HgCl2最可能以分子形式平行吸附在γ-Fe2O3表面。

表5 HgCl2在γ-Fe2 O3表面吸附的优化参数Table 5 Optimized adsorption parameters of HgCl2 adsorbed on γ-Fe2 O3

图7 HgCl2在γ-Fe2 O3表面的优化吸附模型Figure 7 Optimized models of HgCl2 adsorbed on γ-Fe2 O3
2.4 HCl 在γ-Fe2 O3(001)表面对Hg0 的氧化
SCR 催化剂上HCl 催化氧化Hg0一般遵循Eley-Rideal(E-R)机理或Langmuir-HinShelwood(L-H)机制[34,35]。对于L-H 机理,Hg0和HCl 分别吸附在催化剂表面,然后反应生成HgCl2。而对于E-R 机理,一种反应物(Hg0或HCl)首先被吸附在催化剂表面,然后与另一种气态反应物反应生成HgCl2。根据上文吸附结果,HCl 可以通过形成活性氯物种来提高γ-Fe2O3(001)的汞吸附容量,HCl 和Hg0更易共同吸附在γ-Fe2O3(001)表面。然而,γ-Fe2O3(001)表面活性氯物种对汞的非均相氧化需要克服能量障碍才能形成最终产物HgCl2。因此,有必要探索γ-Fe2O3(001)上的非均相氧化过程。根据HCl 和Hg0在γ-Fe2O3(001)表面的化学吸附,提出了γ-Fe2O3催化剂上HCl 氧化Hg0的L-H 机理。
γ-Fe2O3(001)表面HCl 催化氧化Hg0过程中,中间体、过渡态及生成物的构型如图8 所示,相应的反应能量分布如图9 所示。首先,Hg0和HCl 在γ-Fe2O3(001)表面共吸附形成IM1。在IM1 中,Hg0以Fe−Hg 键的形式吸附在Feoct表面。两个HCl 分子在表面解离,生成两个活性Cl 原子。其中一个吸附态Cl 原子逐渐向吸附态的Hg0原子靠近成键:3.047 Å(IM1)→2.709 Å(TS1) →2.385 Å(IM2),而其Cl−Fe 键逐步断裂(2.180 Å(IM1)→3.241 Å(TS1)→4.744 Å(IM2))。过程克服了17.37 kJ/mol 的能垒并放出12.06 kJ/mol 的能量,形成吸附态HgCl。随后,另一个Cl 原子向吸附态HgCl(ads)靠近,HgCl同时逐渐从倾斜变向平行于γ-Fe2O3(001)表面,最终结合形成HgCl2(ads)。随着Cl 和Hg 原子间距的减小,翻越18.72 kJ/mol 能垒,吸附的Hg0最终被氧化成表面键合的HgCl2。
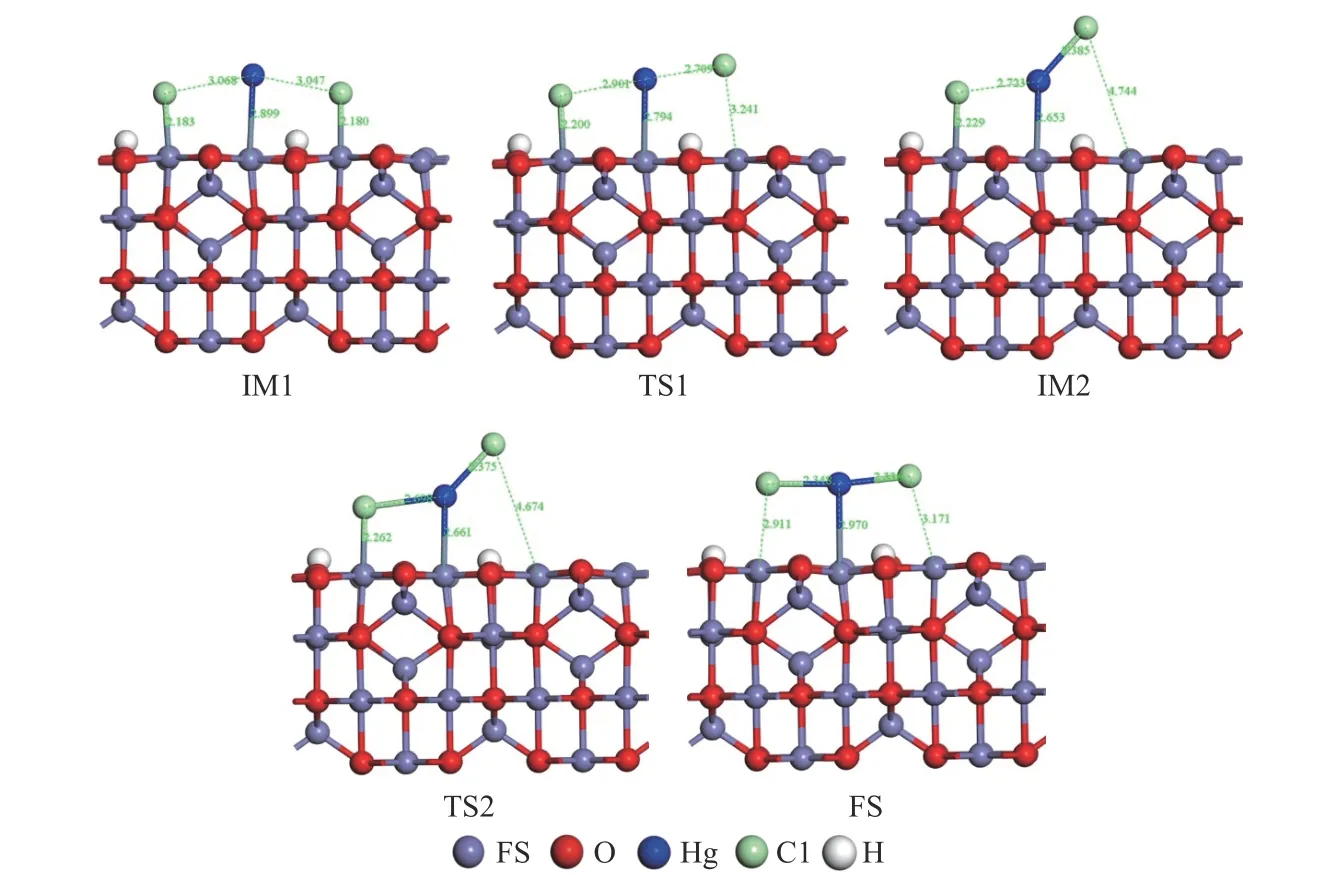
图8 γ-Fe2 O3上HCl 氧化Hg0 的中间体、过渡态和终态的优化结构Figure 8 Optimized structures of intermediate,transition state and final state of Hg0 oxidation by HCl on γ-Fe2 O3
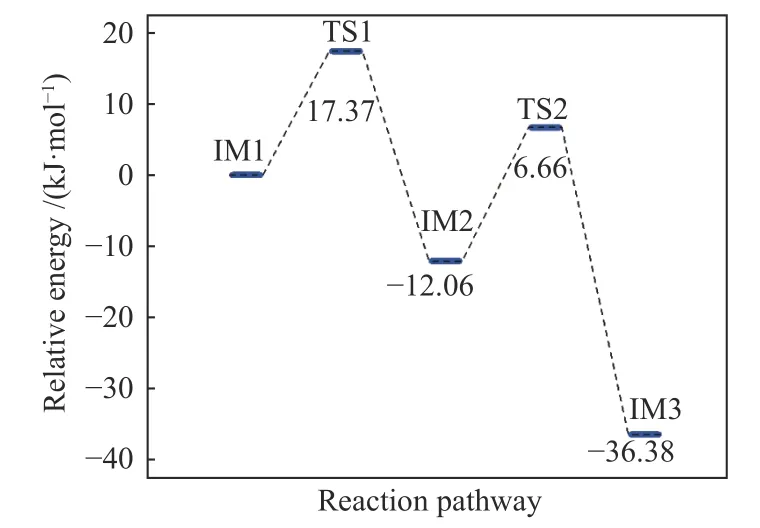
图9 γ-Fe2 O3上HCl 氧化Hg0 的能量分布Figure 9 Energy profile of Hg0 oxidation by HCl on γ-Fe2 O3(001) surface
3 结论
本研究采用密度泛函理论,研究了HCl 在γ-Fe2O3(001)表面催化氧化Hg0的反应机理。发现Hg0化学吸附在γ-Fe2O3催化剂表面的Feoct位,且HCl 在γ-Fe2O3表面的吸附是解离吸附过程。HCl可通过提高邻近原子的吸附活性来促进Hg0在γ-Fe2O3表面的吸附。HgCl 以分子和解离吸附的方式吸附在γ-Fe2O3表面。HgCl2更倾向在γ-Fe2O3表面上以分子形式平行吸附。HCl 在γ-Fe2O3表面的非均相氧化Hg0遵循L-H 机理,即HCl 首先解离吸附在催化剂表面,形成活性的表面氯物种与吸附态的Hg0反应。HCl 在γ-Fe2O3上的非均相氧化汞主要通过两步反应进行,即Hg0(ads)→HgCl(ads)→HgCl2(ads)。
致 谢
本论文的科学计算得到了山东大学的高性能计算云平台的计算支持和帮助。
猜你喜欢
无机化学学报(2023年2期)2023-02-27 03:29:26
建材发展导向(2021年14期)2021-08-23 00:57:14
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08 00:48:08
中国煤层气(2019年2期)2019-08-27 00:59:30
北京航空航天大学学报(2017年10期)2017-04-20 08:51:23
环境与可持续发展(2017年2期)2017-04-06 03:07:30
黑龙江工程学院学报(2016年5期)2016-11-12 05:06:21
物理化学学报(2015年7期)2015-12-30 12:12:50
航天返回与遥感(2014年4期)2014-07-31 17:47:47
火炸药学报(2014年1期)2014-03-20 13:17:27
