
基于SSR分子标记的草果栽培起源分析
2021-12-14 03:41:54李国栋田星赵小丽杨耀文
热带亚热带植物学报 2021年6期
李国栋, 田星, 赵小丽,2, 杨耀文*
基于SSR分子标记的草果栽培起源分析
李国栋1, 田星1, 赵小丽1,2, 杨耀文1*
(1. 云南中医药大学,云南省傣医药与彝医药重点实验室,昆明 650500;2. 德宏州人民医院,云南 潞西 678400)
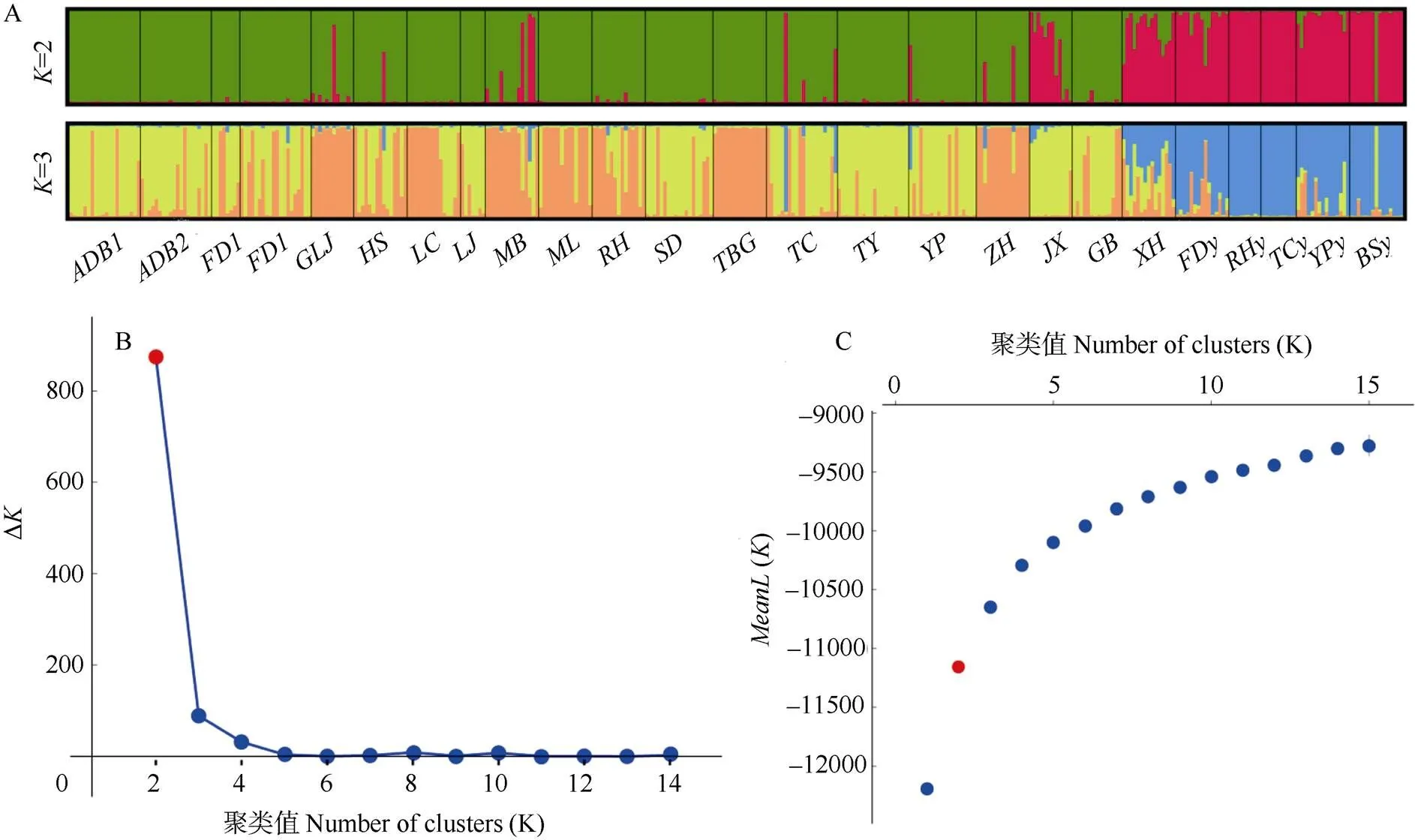
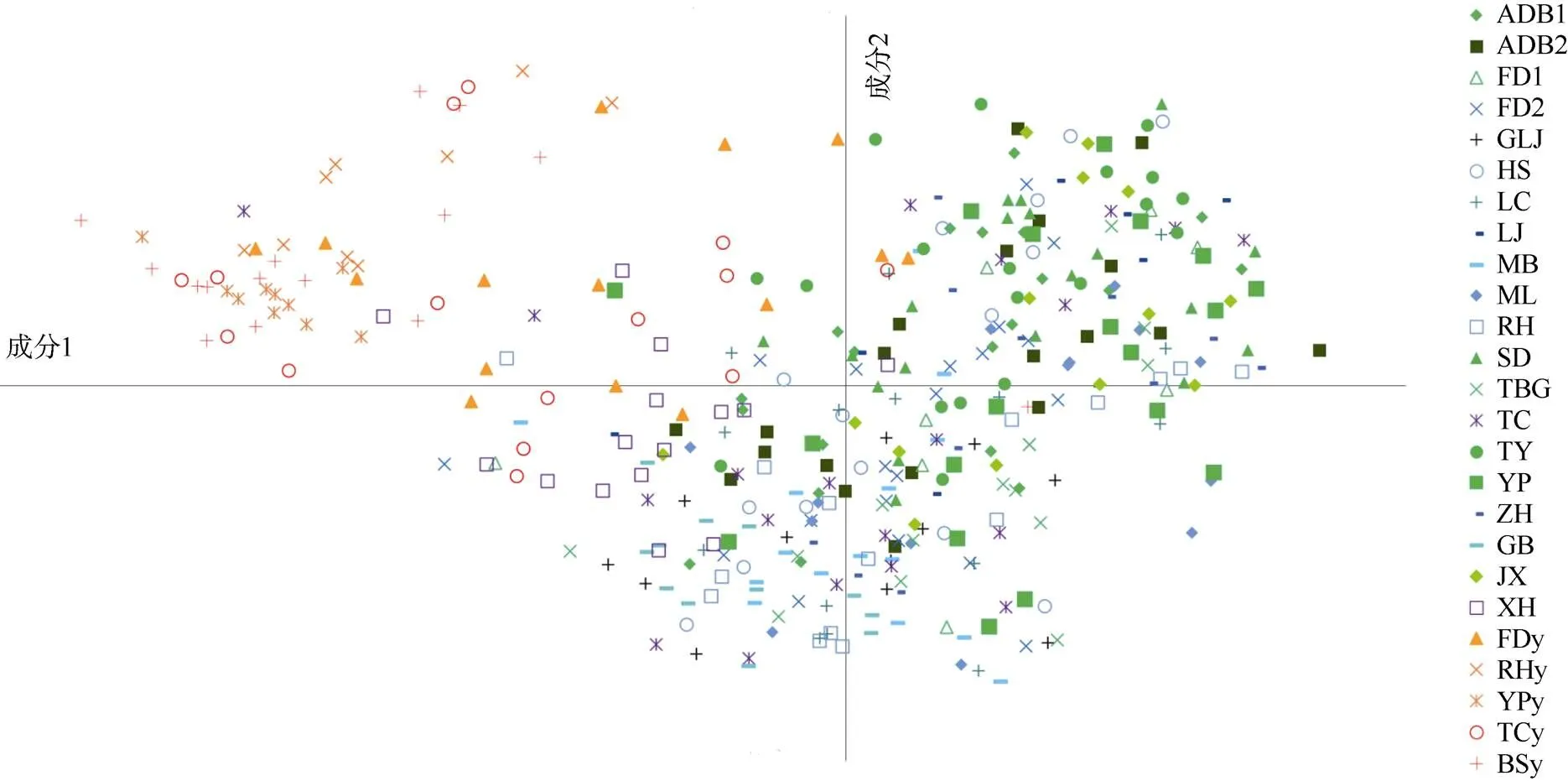
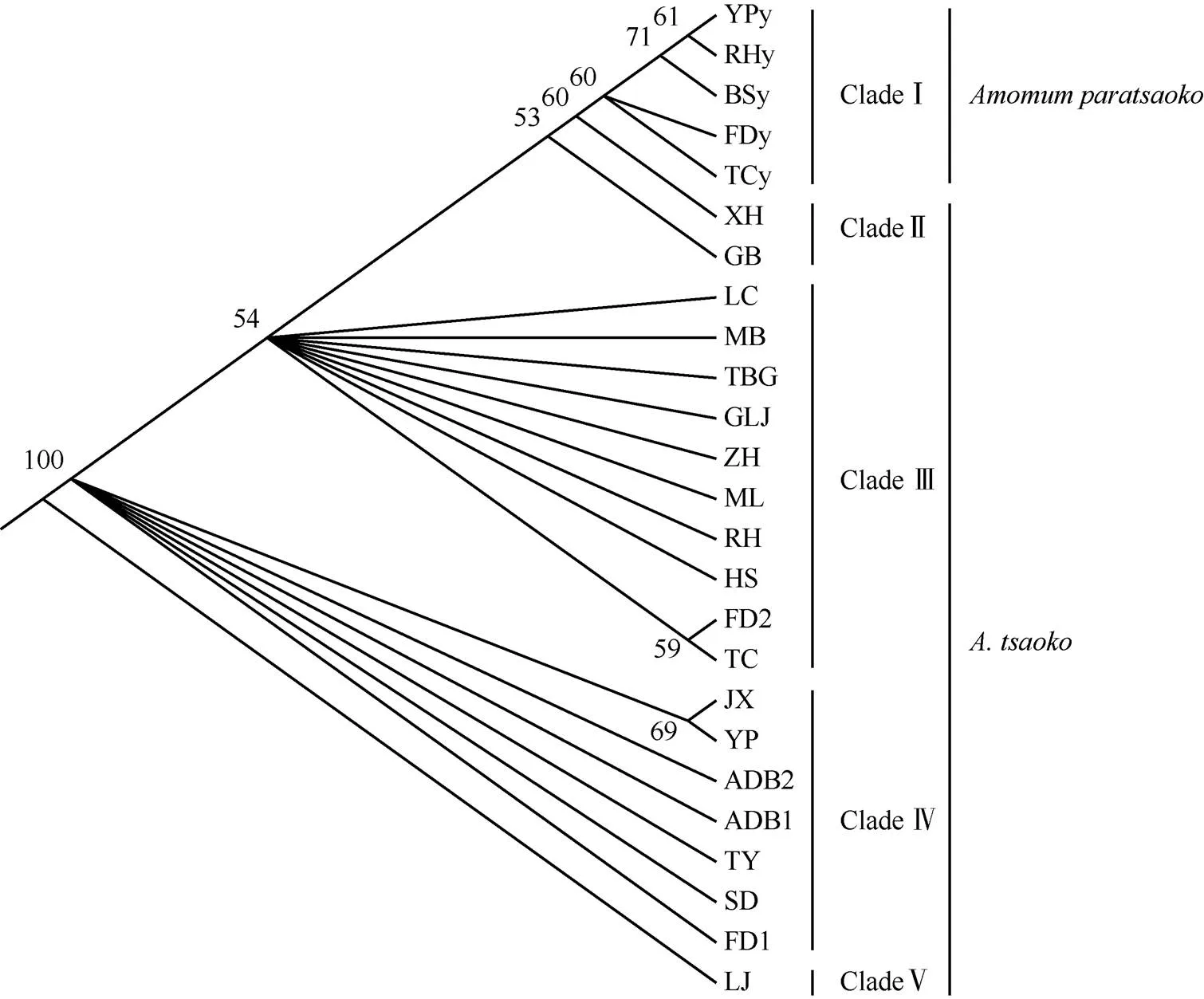
为探索草果()的遗传多样性和栽培起源,对草果和拟草果()在8个SSR位点上的遗传变异进行了分析。结果表明,8个SSR位点在20个草果居群和5个拟草果居群分别检测到149和101个等位基因,特有等位基因分别为44和59个。方差分析(AMOVA)表明,仅10.43%的遗传变异存在于2物种间,8.66%于种内居群间,80.91%于居群内(<0.01)。拟草果居群总的遗传分化程度较大(0.15≤st≤0.25),草果的为中度(0.05≤st≤0.15)。SMM模式下2物种的遗传分化均加大(st 草果;拟草果;SSR分子标记;遗传多样性;栽培起源 草果()主产云南,越南亦分布[1]。果实入药,温中健胃,消食顺气,祛寒湿[2];也作食物辛香料。云南是草果主产区,栽培历史悠久[3]。前期资源调查时在草果传统产区未见姜味砂仁(),仅有野生的拟草果(),且分布邻接区域未见杂交个体。草果、拟草果在生态适应上已有分化:花的行为反应有一定时间差;草果在海拔≥1 200 m林中结果较多,在海拔900 m林中开花多结果极少,而同海拔的拟草果则大量结果;同一海拔,干旱、光照过多或过少,草果生长不好或死亡,而拟草果生长较好,适应性较强; 草果果实不脱落,拟草果果实较草果小,中秋前后脱落、腐烂。这些分化可能是导致2物种无杂交个体的原因。传统草果产区对二者的认识、利用也不同,拟草果(野草果、白草果)仅食用,草果(家草果、红草果)入药, 且种子挥发油成分不同,不能当作同一药材使用[4]。拟草果是与草果亲缘关系最近的同属野生植物[5]。因此, 拟草果的分布、遗传多样性和居群遗传结构均能为草果栽培起源研究提供重要信息。 阐明物种的栽培起源不仅可认识栽培驯化的遗传结果,也利于瞄准优良种质资源提高品质[6]。草果栽培起源是一个迄今未解决的基础问题,本文基于拟草果与草果的亲缘关系,通过比较分析二者的遗传多样性、居群遗传结构,探索草果栽培起源,为其品质提高奠定基础。 试验材料草果()和拟草果()采自中国云南、广西,越南(表1),凭证标本存于云南中医药大学。共20个草果居群和5个拟草果居群,其中TBG居群的种源从越南经缅甸引进,LC、HS、SD、TBG、LJ、TY、ZH和JX居群当地供销社在上世纪从云南文山州、红河州引种栽培,这7居群附近无拟草果分布。每个草果居群随机选取7~26株,株距≥5m;草果居群周边的(直线距离≤200 m)全部拟草果植株作一个居群,悉数采样。采集健康叶片放入含硅胶的密封袋中干燥,于–20℃的冰箱中保存。 表1 材料采集信息 采用改良的植物基因组DNA快速提取试剂盒提取草果和拟草果的总DNA。用本课题组[7]前期筛选的8对多态性位点高的引物进行扩增(表2)。扩增体系总体积为25L,包含DNA聚合酶(1.0 U/L) 12.5L,正反向引物(10mol/L)各1.5L,DNA模板(30 ng/L) 1.0L,用ddH2O补足。扩增程序为94℃预变性4 min,94℃变性40 s;然后64℃退火30~40 s,72℃延伸40 s,共循环33次;最后72℃延伸6 min。产物存于–4℃冰箱。使用1%琼脂糖凝胶进行电泳检测,120 V电泳25~30 min,挑选具明亮条带的PCR产物送上海生工(Sangon Biotech)进行STR检测。 表2 SSR引物信息 采用GENEMARER V 2.4软件统计片段长度,按照GenALEx V 6.4软件[8]格式要求对数据进行整理。用GenALEx V 6.4、Genepop、Fastat、PIC_CALC version软件统计每个位点等位基因的数目(number ofallele,)和特有等位基因数(mumber of unique allele,p)、杂合度、-统计值、遗传多样性和遗传分化系数等参数。遗传分化参数is、st与it为-统计量计算所得,遗传分化参数is、st与it为-统计量基于逐步突变(stepwise mutation model, SMM)模型计算所得。SMM模型下,大小相似的等位基因意味着有更近的亲缘关系;考虑到居群间遗传分化源于基因突变的积累,对于分化历史足够长的居群,基于SMM模型的居群遗传分化检测更为准确[9]。 采用Arlequin软件的分子方差分析(analysis of molecular variance, AMOVA)模块对草果、拟草果居群进行遗传变异来源分布分析。采用STRUCTURE V 2.3进行多位点的贝叶斯聚类分析[10],推断居群最可能的聚类情况()。用Flaush等推荐的混合模型(Admixture model)[11],选择居群间相关的等位基因频率模型(correlated alleles frequencies model);值定义为1~10,每个值设置运行20次。采用Microsatellite Aanlyser V 4.05 (MSA)[12]计算居群间Nei’s遗传距离矩阵,采用GenALEx V 6.4软件进行主坐标分析(principal coordinates analysis, PCoA), 并计算每个居群8个位点的遗传距离,将结果导入POPTREE V 2.0软件[13],根据邻接法(NJ)构建亲缘关系树(Bootstrap=1 000)。 SSR位点的遗传多样性 在20个草果居群中,每个SSR位点的等位基因数为8~34个,平均18.63个,其中At30和At4位点的等位基因最丰富,分别为34和30个。每个位点拥有1~9个特有等位基因,平均5.5个。At4位点具有较高的等位基因丰富度(allelic richness,R)、平均等位基因数(average number of alleles,A)、有效等位基因数(number of effective alleles,E)和Shannon’s信息指数(Shannon’s information index,)值。观测杂合度(observed hetero- zygosity,O)、期望杂合度(expected heterozygosity,E)和无偏差杂合度(unbiased expected heterozygosity,E)分别为0.21~0.73、0.29~0.81和0.30~0.84, 以位点At4和At46的较高。在5个拟草果居群中, 每个SSR位点的等位基因数为8~19个,平均13个,以At4和At5位点的最丰富,分别为19和15个; 每个位点拥有4~11个特有等位基因,平均7.4个; At4位点具有较高的R、A、E和值;O、E和E分别为0.17~0.65、0.35~0.79和0.36~0.83, 以At4位点的较高(表3)。 表3 SSR位点遗传多样性 :等位基因数;P: 特有等位基因数;R:等位基因丰富度;A: 平均等位基因数;E: 有效等位基因数;: Shannon’s信息指数;O: 观测杂合度;E:期望杂合度;E: 无偏期望杂合度。下表同。 :Number of allele;P:Number of unique allele;R: Allelic richness;A: Average number of alleles;E: Number of effective alleles;: Shannon’s information index;O:Observed heterozygosity;E:Expected heterozygosity;E:Unbiased expected heterozygosity. The same is following Table. 居群的遗传多样性 从表4可见,草果和拟草果居群的等位基因分别有34~60和31~53个, 平均为48.55和39.80个,FD2、MB和TCy居群的等位基因数最高,分别为60、60和53个,MB和TCy居群的NP、NA、NE、HO、HE、UHE值较其他居群高。拟草果居群的Np极显著高于草果居群, 从Np来看,拟草果居群遗传多样性极显著高于草果居群。综合p和E、E来看,在拟草果居群中,TCy居群的遗传多样性最高,其次是YPy和Fdy, BSy最低,其基因多样性(gene diversity,d)的变化趋势相同。同样地,在草果居群中,MB居群的遗传多样性最高, 其次是TC居群,接着依次是XH、GB、YP、LC和TBG居群,d的变化趋势基本相同。 从表5可见,25个居群的群体内近交系数(is、is)总体上均大于0,表明与随机自由交配相比, 近交在群体中更加频繁,导致杂合子不足。总体上草果的is小于拟草果,但is大于拟草果,提示草果居群内积累了更多基因突变,遗传分化大于拟草果。草果居群的总体遗传分化指数st和st均为0.05~0.15,表明其遗传分化为中度,拟草果居群的遗传分化较大(0.15 AMOVA分析表明(表6), 仅10.43%的遗传变异存于2物种间,8.66%存在于种内居群间,80.91%于居群内(<0.01)。草果居群的遗传变异有7.1%存在于居群间,92.9%存在于居群内;拟草果居群分别为23.26%和76.74% (<0.01)。拟草果居群间的遗传分化指数(st=0.23)高于草果居群(st=0.07),草果居群的基因流(m=2.20)大于拟草果居群(m=0.85)。 表4 居群的遗传多样性 is: 近交系数;d: 基因多样性。 is: Inbreedingcoefficient;d: Genediversity. 表5 草果(A)、拟草果(B)居群内和居群间的遗传分化 对草果和拟草果所有居群进行贝叶斯聚类分析表明,值的分布结果支持将所有居群分为两组(图1: B, C)。STRUCTURE分析结果表明,=2是比较适合的分组标准(图1: A)。=2时,草果居群以绿色基因池为主,拟草果居群以红色基因池为主。=3时,草果居群以黄色和橙色基因池为主, 拟草果居群以蓝色基因池为主,2物种均无特有基因池(图1: A)。当=15时,草果和拟草果的基因池种类均相同。PCoA分析结果表明(图2),草果个体聚为一组,拟草果个体聚为另一组,两组间是连续的。TC、XH、RH、YP、MB、FD1和FD2居群中部分个体更靠近拟草果,FDy、TCy和BSy居群中部分个体更靠近草果。 表6 基于8个SSR位点的AMOVA分析 图1 基于STRUCTURE的25个居群聚类分析 基于遗传距离构建NJ系统发育树,结果表明,2物种没有表现出完全的单系,呈现一定程度的离散,拟草果居群TCy、FDy、BSy与RHy、YPy聚在一起(图3),20个草果居群分为4组:组II包括GB与XH居群;组III包括LC、MB、TBG、GLJ、ZH、ML、RH、HS、FD2、TC居群;组V包括LJ居群,组IV包括剩余的7个居群,FD2与TC亲缘关系较近;TY、FD1、SD与ADB1、ADB2、LJ亲缘关系较近。 除GB居群外,2物种的E大于O,is均> 0 (表4),说明24个居群均杂合子不足而偏离哈迪-温伯格(HWE)平衡。5个拟草果居群杂合子不足(is=0.15~0.33)的程度低于草果居群(is=0.1~0.46) (表5)。无效等位基因(null allele)可将杂合子标记为纯合子[9],本研究中8个SSR位点均发生杂合子缺乏,无效等位基因不可能在8个位点同时发生;此外,早期研究中,At1、At4、At29、At46和At56位点均未发现无效等位基因[14]。所以,本研究中草果、拟草果居群杂合子不足,不可能是无效等位基因引发的。杂合子不足意味着居群内可能非随机交配[9]。草果、拟草果均有促进杂交的柱头卷曲机制[15–16],但是,温湿度变化对草果柱头卷曲的影响在居群水平上没有完全形成功能上的雌雄异株,影响草果繁育和降低适合度[17]。2物种均缺乏自交不亲和机制[18],自交亲和机制能强化营养繁殖[19];草果繁殖也有老株分化发展新株的方式[15]。这都可能是草果居群杂合子减少的原因。拟草果果实成熟后脱落,不及时采摘即腐烂,这不利于种子远距离扩散,可能是杂合子缺乏的原因,此外,生境受到破坏、样本量少(5个居群63个个体)也可能是原因之一。特有等位基因数(P)、期望杂合度(E)可评价居群遗传多样性的丰富程度[20–21]。据此,我们推测云南马关、麻栗坡、屏边、广西那坡以及越南管箔是草果的遗传多样性中心,而云南麻栗坡、屏边、西畴和马关可能是拟草果的遗传多样性中心,并且提示云南马关、麻栗坡、屏边可能是草果栽培起源地理中心。 图2 草果和拟草果25个居群375个体的PCoA主成分分析 图3 基于遗传距离的草果、拟草果居群NJ系统发育树 在大量野外调查和材料采集中并未发现推定的杂交个体。草果、拟草果共享等位基因占40.45%,这意味着种质渗透,或者是共享祖先的遗传多样性[9]。AMOVA分析结果既支持2物种共享等位基因较多,又与豆蔻属分子系统研究结果一致[5],这表明2物种共享祖先的遗传多样性。 豆蔻属起源于喜马拉雅[22–23],其起源、分化历史较短。草果、拟草果的形态类似、分子系统上亲缘关系较近[1,5]。目前草果和拟草果间无杂交证据, PCoA结果也说明其分化历史短。因为对于共享较多等位基因、在亲缘关系树上混杂分布的物种之间可能发生了种质渗透,也可解释2物种分化历史不久远,无论物种间有无杂交[9]。2物种亲缘关系树也支持草果、拟草果分化历史较短。非SMM和SMM模式皆表明居群分化是由较高的基因流引起的,而对分化历史短的居群,分化主要由随机遗传漂变导致[9]。非SMM和SMM模式下拟草果居群间的遗传分化均大于草果;考虑等位基因长度的SMM分析,遗传分化在种间、同种居群间均加大(表5)。因此,草果、拟草果的分化可能是经随机遗传漂变完成谱系分选后基因突变积累的结果。 当=3时,基因池占比表明XH、TC、MB和YP居群与拟草果的亲缘关系较近(图1: A),可能提示这4个草果居群所处的那坡、麻栗坡、马关、屏边可能是草果栽培起源地理中心。考虑基因流,基于遗传距离构建的NJ树(图3)作为草果居群间亲缘关系的主要依据。5个拟草果居群,马关居群和其西边的屏边居群聚在一起,西畴居群和其东南边的麻栗坡居群、那坡居群聚在一起。西畴、麻栗坡的拟草果居群和草果居群聚在一起,说明与草果的亲缘关系较近,可能提示西畴、麻栗坡是草果栽培起源的1个地理中心。与拟草果居群的亲缘关系较近的6个草果居群(XH、MB、GLJ、ML、FD2和TC),以及与草果亲缘关系较近的3个拟草果居群(FDy、TCy和BSy)所处区域,即麻栗坡、马关、屏边、西畴、那坡可能是草果栽培起源地理中心。20个草果居群分为4组:组II (GB与XH)与拟草果亲缘关系较近,可能代表了草果祖先居群的某些特征;也可能提示云南马关和越南管箔是草果栽培起源地理中心。组III的TBG与GLJ亲缘关系较近,ZH可能是从马关引种的,越南居群可能是马关居群南迁的结果,可能提示马关是草果栽培起源的1个地理中心。私有等位基因可以为作物栽培起源中心的研究提供证据[24]。本研究中MB居群的p最高,其次是YP、GB和TBG居群(表4),提示云南马关、屏边以及越南河江省可能是草果栽培起源地理中心。 综上所述,草果栽培起源的地理中心可能是以云南马关、屏边为中心,向其周边邻近地区扩散而形成的。马关向麻栗坡(铁厂)、那坡(百省)、西畴(法斗)、越南(河江省等)扩散,屏边向金平(阿德博)扩散。YP、MB居群均在大围山范畴,因此,围绕大围山的马关、屏边地区可能是草果栽培起源地理中心。依据《开化府志》, 此区域属于原开化府,麻栗坡、马关等地已有300多年的草果种植历史[25]。拟草果遗传多样性高于草果,特别在居群间遗传分化、遗传多样性方面,可能缘于草果是栽培植物, 经历人工选择后,居群丢失了某些特有的等位基因, 且多数等位基因的分布频率下降,仅存于少数居群中。 [1] WU Z Y, RAVEN P H. Flora of China, Vol. 24 Flagellariaceae through Marantaceae [M]. Beijing: Science Press & St. Louis: Missouri Botanical Garden Press, 2000. [2] Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China, Vol. 1 [M]. Beijing: Chinese Medical Science and Technology Press, 2020: 249–250. 国家药典委员会. 中华人民共和国药典, 一部 [M]. 北京: 中国医药科技出版社, 2020: 249–250. [3] MING J H, HU Y H. Dynamic analysis on production and market of[J]. J Chin Med Mat, 2004, 27(6): 449–451. doi: 10. 3321/j.issn:1001-4454.2004.06.030. 明建鸿, 胡耀华. 草果的产销动态分析 [J]. 中药材, 2004, 27(6): 449–451. doi: 10.3321/j.issn:1001-4454.2004.06.030. [4] XU G J. Species Systematization and Quality Evaluation of Commonly Used Chinese Tiaditional Drugs, Vol. 3 [M]. Fuzhou: Fujian Science and Technology Press, 1999: 39–46. 徐国钧. 常用中药材品种整理和质量研究, 第3册 [M]. 福州: 福建科学技术出版社, 1999: 39–46. [5] XIA Y M, KRESS W J, PRINCE L M. Phylogenetic analyses of(Alpinioideae: Zingiberaceae) using ITS andK DNA sequence data [J]. Syst Bot, 2004, 29(2): 334–344. doi: 10.1600/0363 64404774195520. [6] JONES H, CIVÁŇ P, COCKRAM J, et al. Evolutionary history of barley cultivation in Europe revealed by genetic analysis of extant landraces [J]. BMC Evol Biol, 2011, 11: 320. doi: 10.1186/1471-2148- 11-320. [7] YANG Y W, YANG Z Y, YAN M R, et al. Isolation and character- rization of microsatellite markers for(Zingiberaceae), an economically important plant in China [J]. Genet Mol Res, 2014, 13(4): 8220–8224. doi: 10.4238/2014.October.8.3. [8] PEAKALL R, SMOUSE P E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research [J]. Mol Ecol Not, 2006, 6(1): 288–295. doi: 10.1111/j.1471-8286.2005.01155.x. [9] OLSEN K M, SCHAAL B A. Microsatellite variation in cassava (, Euphorbiaceae) and its wild relatives: Further evidence for a southern Amazonian origin of domestication [J]. Amer J Bot, 2001, 88(1): 131–142. doi: 10.2307/2657133. [10] GOUDET J. FSTAT: A program to estimate and test gene diversities and fixation indices, Version 2.9.3 [EB/OL]. 2001, http://www.unil.ch/ izea/softwares/fstat.html. [11] RITCHARD J K, STEPHENS M, DONNELLY P. Inference of popu- lation structure using multilocus genotype data [J]. Genetics, 2000, 155 (2): 945–959. doi: 10.1093/genetics/155.2.945. [12] FALUSH D, STEPHENS M, PRITCHARD J K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies [J]. Genetics, 2003, 164(4): 1567–1587. doi: 10.1093/ genetics/164.4.1567. [13] TAKEZAKI N, NEI M, TAMURA K. POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface [J]. Mol Biol Evol, 2010, 27(4): 747–752. doi: 10.1093/molbev/msp312. [14] HU Y F, ZHANG X M, XU S Z, et al. Analysis of genetic diversity and genetic relationship ofgermplasm resources in Yun- nan by SSR markers [J]. Chin Trad Herb Drug, 2018, 49(22): 5388– 5395. doi: 10.7501/j.issn.0253-2670.2018.22.025. 胡一凡, 张雪梅, 徐绍忠, 等. 云南草果种质资源的遗传多样性及亲缘关系的SSR分析 [J]. 中草药, 2018, 49(22): 5388–5395. doi: 10. 7501/j.issn.0253-2670.2018.22.025. [15] CUI X L, WEI R C, HUANG R F. Study on the artificial population structure of[J]. SW China J Agric Sci, 1995, 8(4): 114–118. 崔晓龙, 魏蓉城, 黄瑞复. 草果人工种群结构研究 [J]. 西南农业学报, 1995, 8(4): 114–118. [16] LI Q J, XU Z F, KRESS W J, et al. Flexible style that encourages outcrossing [J]. Nature, 2001, 41(6827): 432. doi: 10.1038/35068635. [17] YANG Y W, QIAN Z G, LI A R, et al. The influence of temperature and humidity on stylar curvature in(Zingiberaceae) [J]. Plant Diver Resour, 2013, 35(5): 613–620. doi: 10.7677/ynzwyj2013 12171. 杨耀文, 钱子刚, 李爱蓉, 等. 温度和湿度对草果花柱卷曲的影响 [J]. 植物分类与资源学报, 2013, 35(5) 613–620. doi: 10.7677/ynzwyj 201312171. [18] CUI X L, WEI R C, HUANG R F. A preliminary study on the genetic system of[J]. J Yunnan Univ (Nat Sci), 1995, 17(3): 290–297. 崔晓龙, 魏蓉城, 黄瑞复. 草果遗传体系的初步研究 [J]. 云南大学学报(自然科学版), 1995, 17(3): 290–297. [19] DELPLANCKE M, YAZBEK M, ARRIGO N, et al. Combining conservative and variable markers to infer the evolutionary history ofsubgen.s.l. under domestication [J]. Genet Resour Crop Evol, 2016, 63: 221–234. doi: 10.1007/s10722-015-0242-6. [20] PAN Y Z, WANG X Q, SUN G L, et al. Application of RAD sequencing for evaluating the genetic diversity of domesticated(Araliaceae) [J]. PLoS ONE, 2016, 11(11): e0166419. doi: 10.1371/journal.pone.0166419. [21] MEEGAHAKUMBURA M K, WAMBULWA M C, THAPA K K, et al. Indications for three independent domestication events for the tea plant [(L.) O. Kuntze] and new insights into the origin of tea germplasm in China and India revealed by nuclear microsatellites [J]. PLoS ONE, 2016, 11(5): e0155369. doi: 10.1371/journal.pone.015 5369. [22] KAM Y K. The genus Elettariopsis (Zingiberaceae) in Malaya [J]. Notes RBG Edinb, 1982, 40(1): 139–152. [23] SMITH R M. A review of Bornean Zingiberaceae I: (Alpineae) [J]. Notes RBG Edinb, 1985, 42(2): 261–314. [24] GUO Y M, CHEN S, LI Z Y, et al. Center of origin and centers of diversity in an ancient crop,(Turnip Rape) [J]. J Hered, 2014, 105(4): 555–565. doi: 10.1093/jhered/esu021. [25] TANG D B, ZHOU B. Annotation of “Annals of Kaihua Prefecture” [M]. Lanzhou: University of Lanzhou Press, 2004: 111. 汤大宾, 周炳. 开化府志点注 [M]. 兰州: 兰州大学出版社, 2004: 111. Analysis of Cultivation Origin ofBased on SSR Marker LI Guodong1, TIAN Xing1, ZHAO Xiaoli1,2, YANG Yaowen1* (1. Yunnan Key Laboratory of Dai and Yi Medicines, Yunnan University of Chinese Medicine,Kunming 650500, China; 2. Dehong People’s Hospital,Luxi 678400, Yunnan, China) In order to explore the genetic diversity and geographical origin of, the genetic variation ofandin 8 SSR loci was studied. The results showed that there was 149 and 101 alleles were detected at 8 SSR loci from 20 populationsofand 5 populations ofrespectively, among which 44, 59 were unique alleles. Variance analysis (AMOVA) showed that only 10.43% of genetic variation existed between two species, 8.66% between populations, and 80.91% within populations (<0.01). The total genetic differentiation ofwas moderate (0.05≤st≤0.15), andwas high (0.15≤st≤0.25). The genetic differentiation of the two species increased under SMM model (st ;; SSR marker; Genetic diversity; Origin of cultivation 10.11926/jtsb.4377 2021-01-12 2021-04-30 云南省应用基础研究-中医联合项目(2018FF001-010, 2017FF116-004); 国家自然科学基金项目(81660631); 重大科技专项(2018ZF010-1)资助 This work was supported by the Cooperation Project of Traditional Chinese Medicine and Application Fundamental Research in Yunnan (Grant No. 2018FF001-010, 2017FF116-004), the National Natural Science Foundation of China (Grant No. 81660631), and the Special Project for Major Science and Technology (Grant No. 2018ZF010-1). 李国栋(1984~ ),男,博士,副教授,主要从事中药资源学与分子生药学的教学与研究。E-mail: gammar116@163.com 通信作者 Corresponding author. E-mail: yangyaowen@ynutcm.edu.cn1 材料和方法
1.1 材料
1.2 总DNA的提取和PCR扩增

1.3 数据分析
2 结果和分析
2.1 遗传多样性
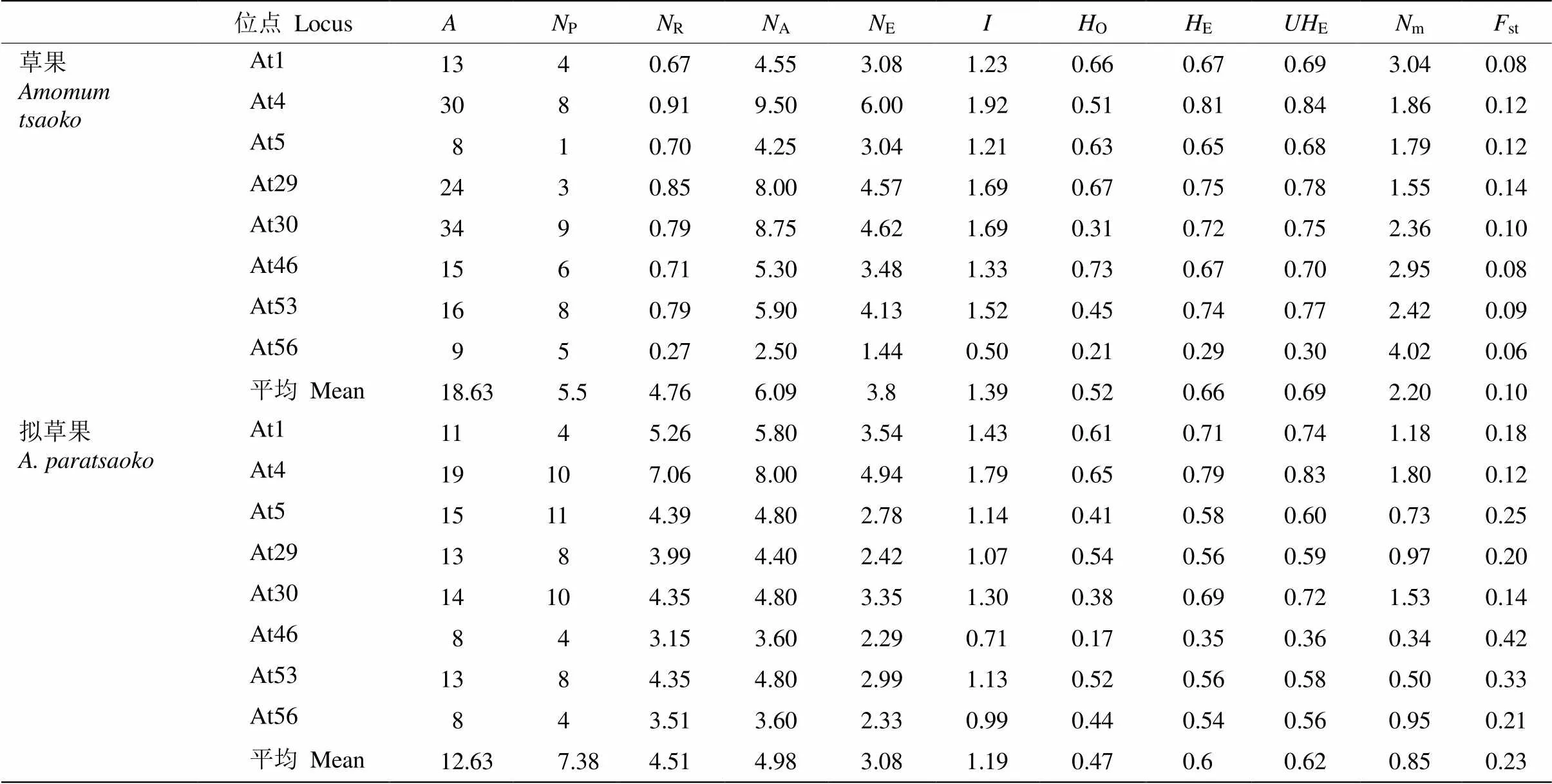
2.2 遗传结构
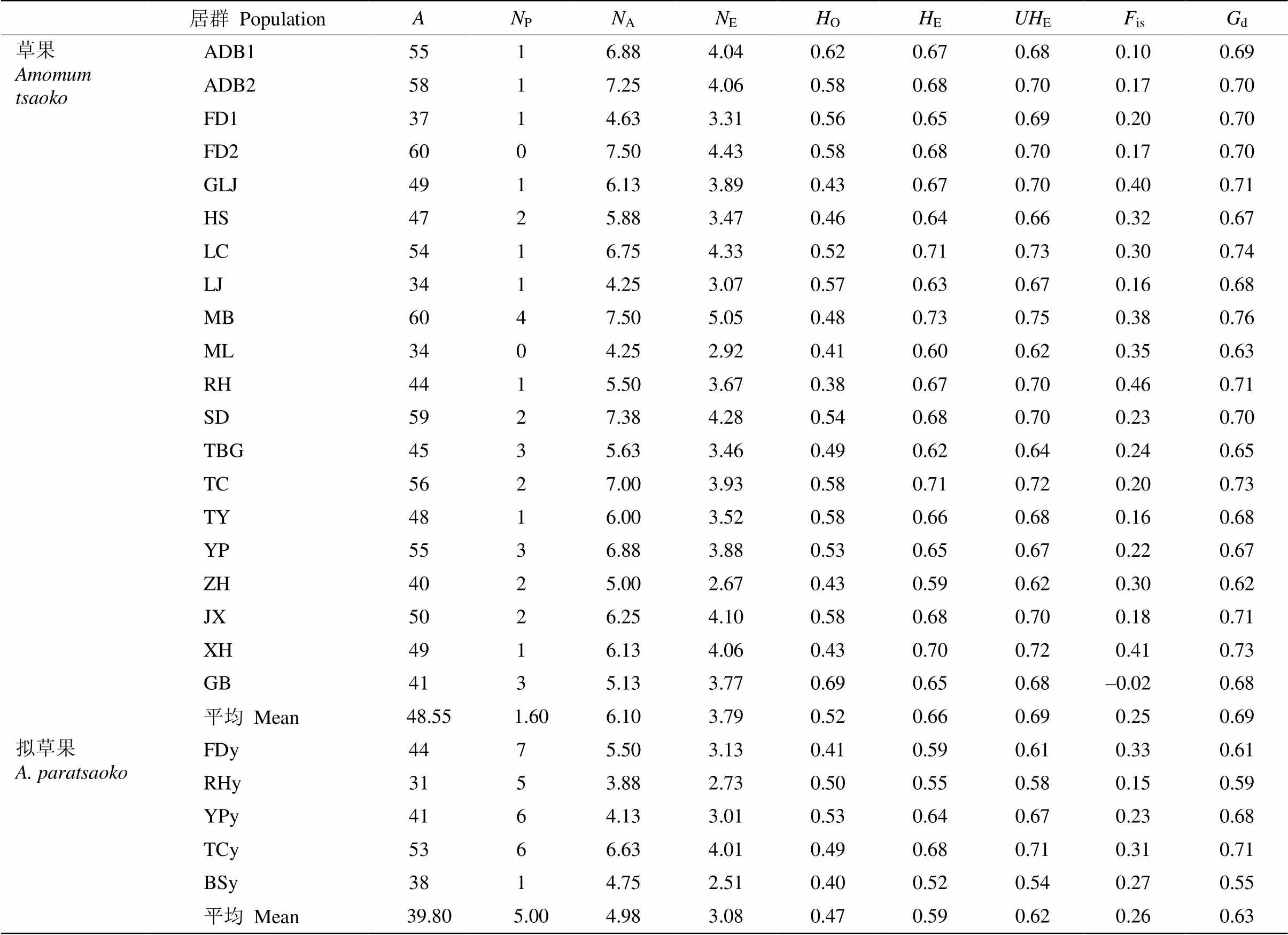

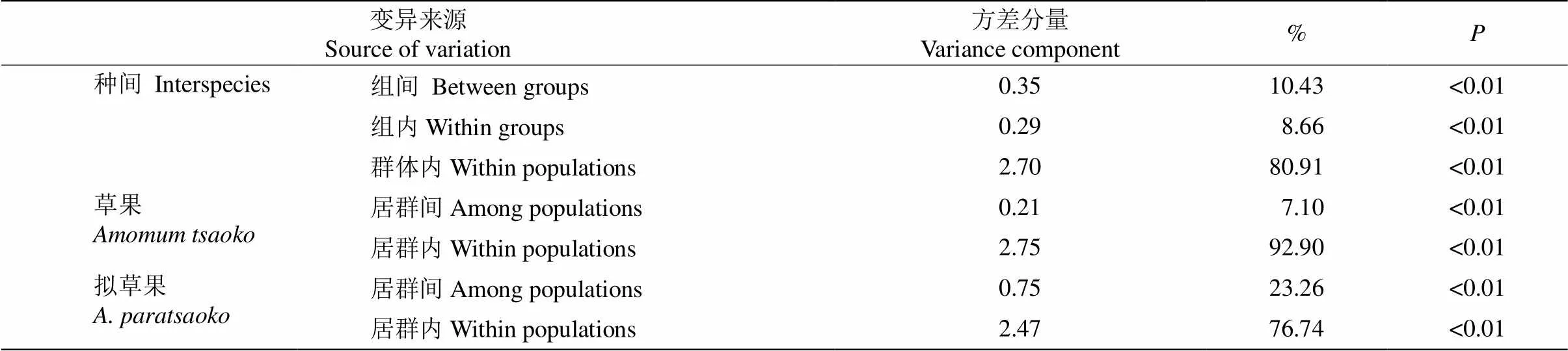
2.3 亲缘关系
3 结论和讨论
 猜你喜欢
河北科技师范学院学报(2022年2期)2022-08-26 08:55:24食品安全导刊(2021年21期)2021-08-30 08:21:26浙江中医药大学学报(2021年6期)2021-07-12 03:05:14音乐天地(音乐创作版)(2019年6期)2019-09-20 05:53:56散文(2019年6期)2019-09-10 07:22:44草地学报(2018年5期)2018-11-07 02:25:00中央民族大学学报(自然科学版)(2016年1期)2016-06-27 07:54:20云南地质(2015年3期)2015-12-08 06:59:56含笑花(2014年6期)2015-01-05 02:43:44含笑花(2012年6期)2012-04-29 02:38:38
猜你喜欢
河北科技师范学院学报(2022年2期)2022-08-26 08:55:24食品安全导刊(2021年21期)2021-08-30 08:21:26浙江中医药大学学报(2021年6期)2021-07-12 03:05:14音乐天地(音乐创作版)(2019年6期)2019-09-20 05:53:56散文(2019年6期)2019-09-10 07:22:44草地学报(2018年5期)2018-11-07 02:25:00中央民族大学学报(自然科学版)(2016年1期)2016-06-27 07:54:20云南地质(2015年3期)2015-12-08 06:59:56含笑花(2014年6期)2015-01-05 02:43:44含笑花(2012年6期)2012-04-29 02:38:38
