
转动光谱研究分子间σ-hole和π-hole非共价相互作用
2021-12-14 14:53陈军华王浩郑阳程琬滢李卫星徐雪芳勾茜
量子电子学报 2021年6期
陈军华,王浩,郑阳,程琬滢,李卫星,徐雪芳,勾茜,3*
(1重庆大学化学化工学院,重庆 401331;2复旦大学化学系,上海 200438;3重庆理论与计算化学重点实验室,重庆 401331)
0 引言
化学科学在上世纪的发展使人们的生活和生产发生了革命性变化,如基于石油化工的能源燃料及高分子材料等,因此有学者将20世纪化学科学的发展归结为对共价键的认识。然而,人类生活水平提高的同时也面临着能源及环境带来的严峻挑战,如何解决这些问题也成为当代人刻不容缓的使命。高效绿色化学反应和可持续性新型材料的设计是解决以上问题的关键。分子间非共价键在这些过程中扮演至关重要的角色,是解决这些问题的关键所在,因此非共价键被誉为本世纪化学领域研究的核心[1]。近年来,呈指数级增长的非共价相互作用的理论[2,3]和实验[4-6]研究报道也间接印证了这一观点。此外,生物特异性识别[7]、超分子自组装[8]和催化[9,10]等过程同样是通过分子间非共价相互作用实现的。因此,以分子复合物为研究模型,从结构和能量上认识分子间非共价键相互作用的方式、作用位点、构象平衡及内部动力学过程,能够准确地揭示宏观生物活性、晶体形态及化学转化等过程的物理本源,也促使新型药物、催化剂、小分子生物酶、超分子配合物等的精准设计成为可能。
随着对非共价键研究的不断深入,从范德华力到氢键,再到卤键,以及近年来发现的碳族、氮族和氧族的原子作为电子受体参与形成的非共价相互作用,对非共价相互作用本质的认识越来越全面。在探讨卤键本质的过程中,Clark等[11]基于分子表面静电势提出了一个物理意义更加明确的概念,即“σ-hole”,它是指沿着正向极化的σ共价键的键轴方向延伸的原子外围中心正表面静电势或低电子密度区域。以卤键R-X···Y为例(X为卤素原子,Y表示卤键受体),σ-hole位于卤素原子X的共价键R-X反方向的正中心。根据σ-hole的定义,有人把氢键也归类为σ-hole非共价相互作用范畴[12,13]。类似地,处于正静电势或低电子密度区域的π电子体系,则被称为“σ-hole”,其作用方向一般垂直于分子的骨架结构。研究发现含碳族、氮族和氧族原子的分子不仅能形成π-hole非共价相互作用,也可以形成π-hole非共价相互作用。
量子化学计算可以预测非共价相互作用的成键类型、能量、结构及内部动力学过程等信息,进而用来指导实验数据的解析[14,15]。近年来开发的密度泛函方法结合色散力等高阶弱相互作用矫正新方法,可以在合理的计算时间内得到较为准确的大尺寸分子或团簇的结构及能量[16,17],通过高水平理论计算,一些新的非共价相互作用类型,如 N···O、N···N、C···C 和 O···O 等,逐步被预测并在团簇稳定性中起决定性作用。这些发现引起了实验光谱学者的注意[18,19],为进一步实验研究提供了方向和理论指导。对于一些复杂的分子体系,尤其是由弱非共价相互作用结合的大尺寸分子团簇,目前的量化计算水平还无法给出精确的结构及相对能量[20]。因此,高精度、高分辨的实验方法反过来成为新计算方法的试金石。X射线衍射[21,22]、红外光谱[23,24]、红外光解离光谱技术[25,26]、拉曼光谱[27,28]以及核磁共振[29,30]等均可用于非共价相互作用的研究,虽然红外和拉曼光谱也可以用于气相研究,但是这些实验方法以凝聚相为主,往往受到分子所处环境中的基质效应[31]、晶体效应[32]和聚集效应[33]的影响,从而无法从本质上研究各类非共价相互作用。光谱技术结合超声射流冷却技术可以有效地排除外部其他分子及作用力的干扰,是研究非共价相互作用本质的有效手段。特别是高分辨转动光谱技术研究分子体系转动能级间的跃迁,能够区分体系不同的构型或构象及非共价相互作用模式[34-37],并获得准确的结构、能量及内部动力学过程等信息。转动光谱原则上可以在不依托理论计算预测情况下获得对象分子的结构参数及内部动力学等信息,这是其他实验方法难以实现的[4,6,20,38]。
本文将简单介绍转动光谱的基本原理及其研究非共价相互作用的优势,重点综述近年来转动光谱研究σ-hole和π-hole非共价相互作用方面的进展。
1 转动光谱基本原理
基于玻恩-奥本海默近似,分子内部运动可以拆分为电子相对原子核的运动、原子核之间的相对振动、分子本身的转动以及电子和原子核的自旋。在分子光谱中,分子与电磁辐射作用可以分别产生电子跃迁(主要在紫外可见光频段)、振动跃迁(主要在红外频段)及转动跃迁(主要在微波频段,因此也常被称为微波光谱)。如图1所示,分子的转动能级差(对于大多数分子0.01~100 cm-1)远小于其电子及振动能级差。分子转动也可以与分子内部其他运动(如原子核自旋、核量子隧穿、电子运动等)耦合,从而产生超精细结构。因此分子转动光谱具有超高能量分辨,能够提供分子精确的结构、电荷分布、内部动力学等准确信息。转动哈密顿算符与分子转动惯量直接相关,故转动光谱能够精确辨识分子在结构上的微小差异(如不同的构象、异构体、同位素取代物等)。利用此优势,微波光谱可以用来确定未知分子种类及结构,如探测星际分子、研究分子间弱相互作用。虽然微波光谱具有许多不可取代的优势,但是转动跃迁选律要求分子必须具有永久电偶极矩(或磁偶极矩,如三重态的氧分子[39])。因此对于大多数高对称分子,由于没有永久电偶极矩,无法进行微波光谱研究。但是可以利用一些“小窍门”来克服这一弊端,如不对称同位素取代法,利用不同同位素在分子中的振动差异创造出偶极矩[40]。
转动光谱研究最早可追溯到20世纪30年代[41-43],Cleeton和Williams在1934年使用吸收式光谱仪首次完成了NH3分子的转动光谱研究[43]。吸收式微波光谱仪主要包括微波源、波导样品池、超外差接收器三部分,通过测量经过样品池的微波辐射强度获得样品分子的微波吸收频谱。20世纪50年代早期,微波光谱领域开始利用样品发出的瞬时自由感应衰减信号来提高转动光谱检测的灵敏度。1979年,Balle和Flygare基于腔增强技术,将微波脉冲与Fabry-P´erot谐振腔结合[44,45],设计了Balle-Flygare傅里叶变换微波(BF-FTMW)光谱仪。BF-FTMW光谱仪结合超声射流冷却技术,使进入共振腔的分子或团簇经绝热膨胀快速冷却至几K,大部分分子布居在振动基态,大大提高了转动光谱的灵敏度和分辨率。已搭建的主流BF-FTMW光谱仪测量范围一般为6 GHz以上,部分原因是由于谐振腔尺寸及微波传输效率的限制,工作频率范围难以扩展到更低的频段,最主要的因素是绝大多数振动基态分子的转动跃迁在6~18 GHz频率范围内都有很强的光谱信号。由于其工作范围超过10 GHz,之前基于Fabry-P´erot谐振腔的FTMW光谱仪被称为“宽带”傅里叶变换微波光谱,但是其单次测量的带宽只有1 MHz左右,扫描1 GHz频率范围的转动光谱需要几个小时。随着微波和电子元器件技术的发展,2008年,弗吉尼亚大学的Pate课题组[46]设计了真正意义上的“宽带”啁啾脉冲傅里叶变换微波(CP-FTMW)光谱仪。他利用任意波形发生器来产生一个线性扫频微波脉冲,可在几微秒时间内扫描10个GHz的频率范围。由于缺少Fabry-P´erot谐振腔来增强微波辐射,因此需要一个高功率放大器稳定放大微波输入信号(几百瓦甚至是上千瓦)。另外不使用差频技术的情况下,则需要一个高采集速率的示波器收集分子的发射信号。虽然CP-FTMW大大提高了信号采集效率,但一般而言,CP-FTMW光谱仪难以媲美BF-FTMW光谱仪的灵敏度和分辨率,并且其造价是后者的几倍。
基于Fabry-P´erot谐振腔以及宽带啁啾(chirp)脉冲的傅里叶变化微波光谱仪是目前的主流设计。图2是重庆大学设计搭建的分子束-共振同轴-傅里叶变换微波光谱仪的结构示意图,主要包括Fabry-P´erot真空谐振腔、真空泵系统、微波信号产生及控制系统、脉冲电磁阀、信号采集系统、反射镜面运动控制系统、时序控制系统等。该仪器的工作频率范围为2~20 GHz,可根据需要扩展至20~60 GHz;分辨率达到3 kHz,频率测量精度达1 kHz。该FTMW光谱仪采集转动光谱信号的基本工作原理如图3所示:1)目标分子以脉冲形式(~0.5 ms)随载气经超声喷射进入真空谐振腔形成低温分子束,实现分子束的在线制备;2)特定频率的微波脉冲(0.2~1.0 μs)进入真空谐振腔使样品发生极化;3)微波辐射停止后,分子或团簇发生弛豫,产生自由感应衰减瞬态信号,被检测器接收(~300 μs),经快速傅里叶变换得到频域谱图。由于分子束-微波同轴传输产生的多普勒效应,每条转动跃迁的谱线将成对出现,分子的跃迁频率为两条多普勒谱线频率的平均值。成对出现的谱线不仅可以排除电子元件自身产生的干扰信号,还提高了光谱分辨率及频率和精确度。
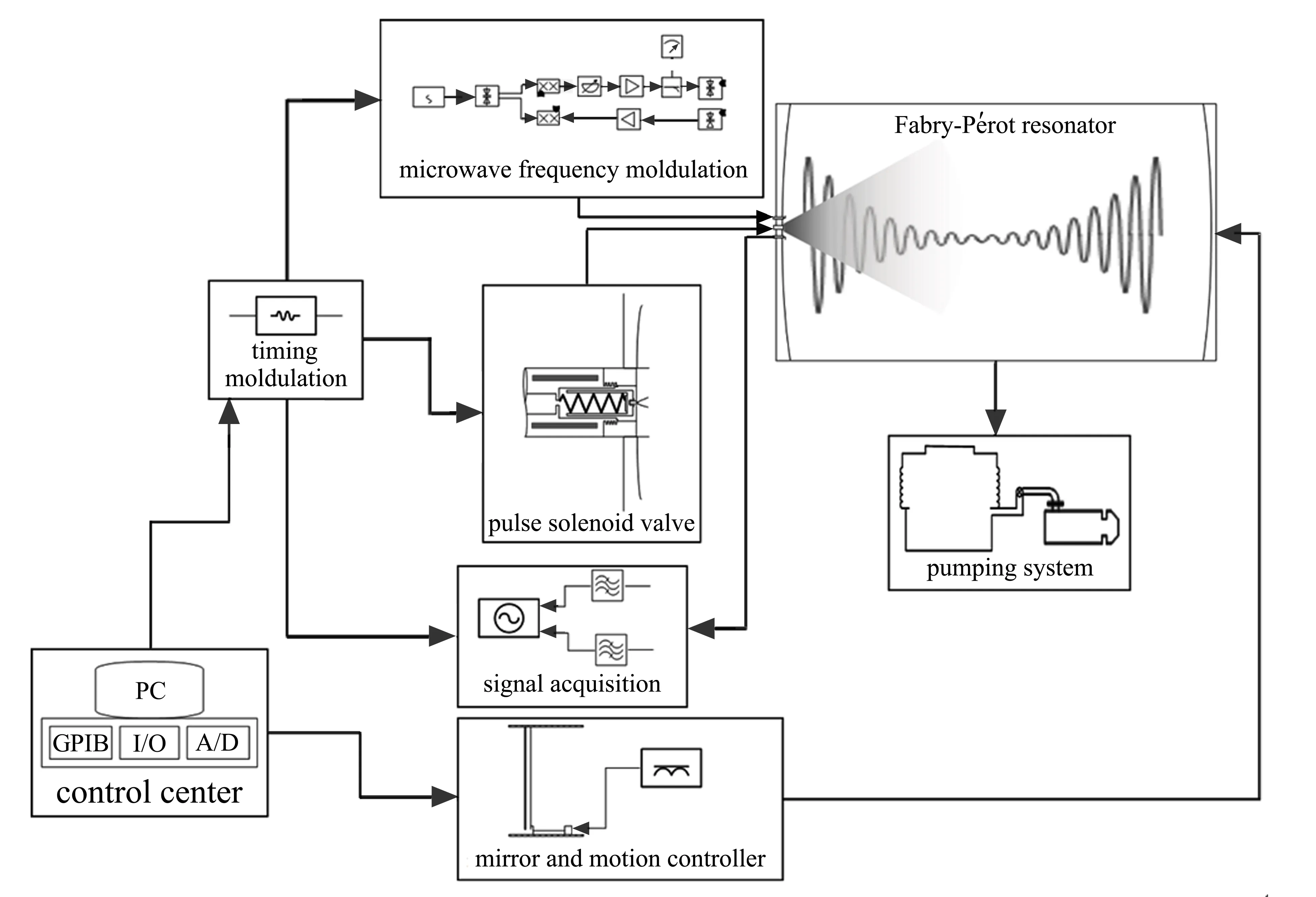
图2 同轴分子束-傅里叶变换微波谱仪结构图[47]Fig.2 Construction of the coaxially oriented beam-resonator Fourier transform microwave spectrometer[47]

图3 傅里叶变换微波谱仪工作原理简图。分子脉冲通过电磁脉冲阀超声射流进入真空谐振腔,然后特定频率的微波脉冲进入真空谐振腔使样品发生极化,停止微波辐射后接收自由感应衰减的时域信号,经过傅里叶变换得到频域转动光谱[38]Fig.3 Operating principle diagram of Fourier transform microwave spectrometer.The molecular supersonic pulse is expanded into the vacuum resonator through a solenoid valve,then a microwave pulse with specific frequency is pulsed into the vacuum resonator and polarize the sample.Once microwave radiation stops,the free induction delay is recorded in time domain and converted to rotational spectrum of frequency domain through Fourier transformation[38]
2 σ-hole非共价相互作用
σ-hole是原子形成的共价键反方向的最外表面处正的静电势或低电子密度区域,在σ-hole的概念提出之前,Brinck等[48]在1992年就已经通过理论计算发现卤素原子上存在正电势区域,并且能够与分子的富电子位点相互作用,形成现在所谓的卤键。随着对σ-hole的深入研究,发现第VI、V和IV主族的原子也能形成σ-hole[49,50]。现在,部分研究人员将氢键[51,52]也归类为σ-hole作用。关于氢键的转动光谱研究已经非常广泛,而且已有相应的综述论文[32,53],故此处仅讨论目前关注度比较高的卤素、氧族及碳族原子形成的σ-hole非共价相互作用的转动光谱研究。对于氮族σ-hole非共价相互作用的转动光谱研究非常有限,仅在F3P-H2O复合物[54]中观察到了以P原子作为σ-hole供体的P···O相互作用。
2.1 卤键
关于σ-hole卤键的转动光谱起始于Legon对一系列双卤原子分子与小分子复合物的研究,其中涉及到的双卤原子分子包括F2[55-59]、Cl2[60-64]、Br2[65]、ClF[55,66,67]、BrCl[68,69]和ICl[70-79]与小分子如H2O、NH3、CO、CH≡CH、CH2=CH2等形成的卤键复合物。通过转动光谱数据对这类卤键作用的分子间/内的电荷转移、伸缩力常数等进行了深入分析和总结[80,81],结果表明:F、Cl、Br和I原子均可以作为卤键的供体,并且异核双卤原子分子与小分子形成卤键时均是周期数较大的卤素原子作为卤键供体。
近十年来,对卤键的研究逐渐扩大到分子尺寸更大的全卤代烷烃分子,其中,CF3Cl和CF3I是最常见的模型分子。在CF3Cl和CF3I分子中,由于-CF3的强吸电子性,在C-Cl/I键反方向的氯/碘原子外表面均形成了一个明显的σ-hole,这很容易与分子的富电子位点相互作用形成卤键。CF3Cl与H2O[82]、H2CO[83]、CH3OCH3[84]的复合物均是以C-Cl···O卤键作用结合在一起的。因为Cl(35Cl和37Cl)的核自旋量子数I=3/2,使得每一个转动跃迁均显示出Cl的核四极耦合效应的超精细结构(当分子体系中有原子的核自旋量子数I≥1时,原子的核自旋角动量会与分子整体转动角动量发生耦合,使分子的转动能级发生裂分,从而在转动光谱中显示出核四极耦合效应的超精细结构)。如图4所示为CF3Cl-H2CO[82]复合物505←404跃迁的转动光谱图,由于甲醛绕其C2轴的内转动,使得跃迁裂分为υ=0和υ=1两条谱线,每条裂分均显示出35Cl的核四极耦合效应的超精细结构;类似地,37Cl同位素异形体同样显示出甲醛内转动的裂分和37Cl的核四极耦合效应的超精细结构。尽管核四极的超精细结构使得转动光谱的条数变得更多,但这种特有的裂分模式通常可以作为解析转动光谱的一个切入点。在CF3Cl-NH3[85]/吡啶[86]两个复合物中观察到了C-Cl···N卤键的存在。同样,由于N的核自旋量子数I=1,会使转动光谱进一步发生14N的核四极耦合效应的裂分,使得光谱解析更加复杂。此外,除了与电负性强的O和N原子形成卤键外,卤原子之间也能够以卤键的形式结合,比如在CF3Cl-CH3F[87]复合物中观察到的C-Cl···F相互作用。CF3Cl-CO[88]复合物则以C-Cl···C卤键稳定结合,这是因为尽管碳的电负性小于氧,但反馈π键(π-backdonation)的作用使得CO中C原子带负电。CF3Cl复合物的转动光谱通常表现出明显的内部动力学特征,比如,虽然量子化学计算表明CF3Cl-H2O[83]/NH3[85]是典型的不对称转子,但由于-CF3的大振幅运动,两个复合物的转动光谱均具有对称转子的特征;而与H2CO[82]/CH3F[87]/CH3OCH3[84]/吡啶[86]等形成复合物时,由于-CF3绕其对称轴的低能垒内转动(<10 cal/mol),使得转动常数A远远大于理论计算的预测值。类似地,CF3I与小分子形成的CF3I-CO[89]、CF3I-N2[90]/NH3[91]/N(CH3)3[91]、CF3I-H2O/H2S[92]的复合物分别以 C-I···C、C-I···N、C-I···O/S 卤键相互作用。此外,在 CF3I-CH2=CH2[93]复合物中观察到了π电子体系作为卤键受体的相互作用。全卤化的乙烷分子CF3CF2Cl与H2O也是通过C-Cl···O卤键相互作用[38],相比于CF3Cl-H2O体系,CF3取代CF3Cl其中的一个F原子没有改变复合物的作用方式(C-Cl···O卤键),但CF3的电负性略小于F原子,CF3CF2Cl-H2O复合物的解离能也有所降低。
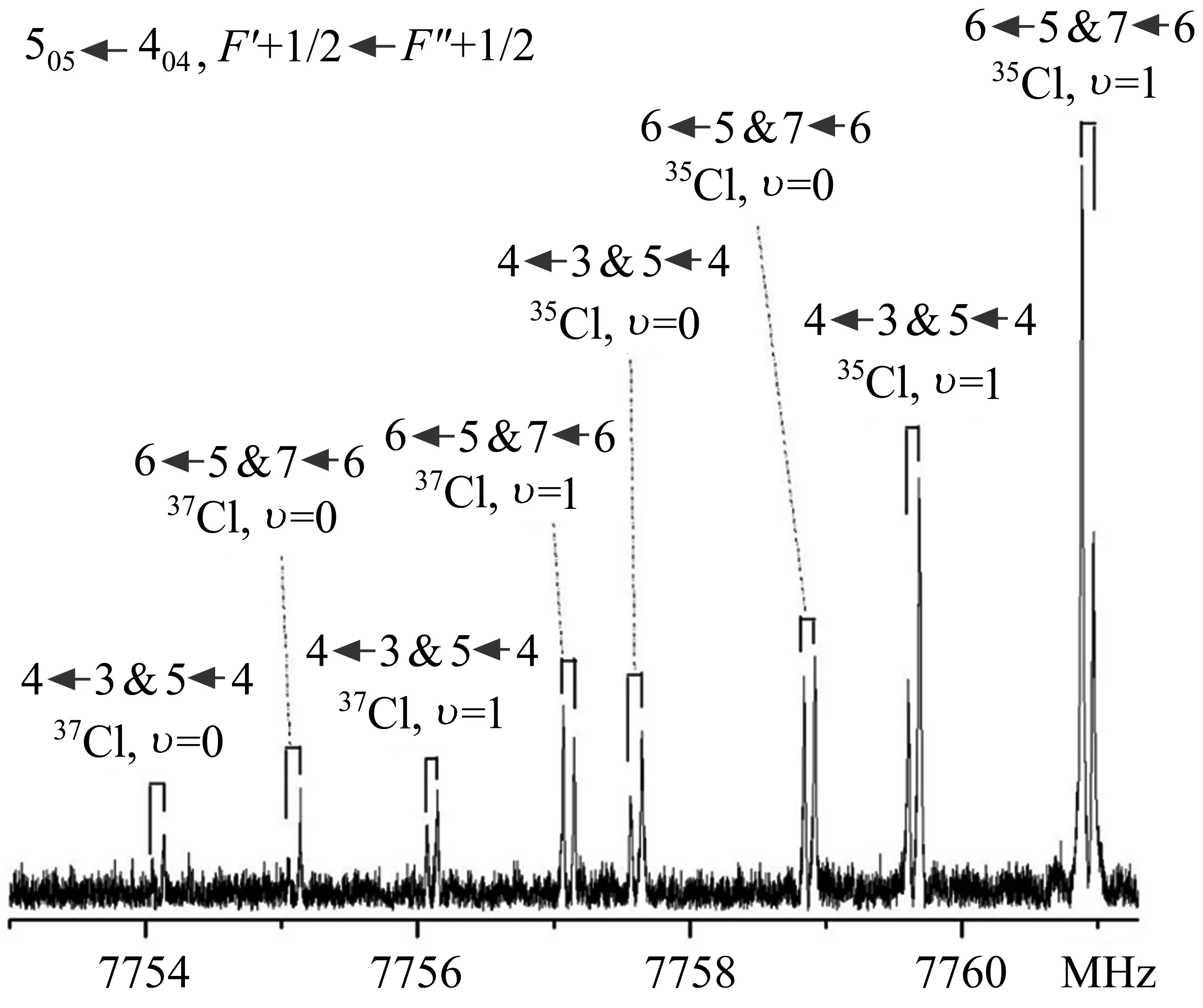
图4 CF3Cl-H2CO复合物35Cl和37Cl同位素异形体505←404跃迁的转动光谱图,每条谱线均显示出多普勒分裂(⊓)[82]Fig.4 Rotational spectrum of the 505←404rotational transition of35Cl and37Cl isotopologues of CF3Cl-H2CO complex,each component line exhibits an instrumental Doppler doubling(⊓)[82]
图 5 对比了 CF3Cl、CF3I与不同小分子形成的 C-Cl/I···C、C-Cl/I···N、C-Cl···O/S 卤键的伸缩力常数Ks,其大小与卤键强度成正比。CF3Cl和CF3I与相同小分子形成卤键时,I原子作为卤键供体的强度明显大于对应Cl原子形成的卤键,表明质量越大的卤素原子形成的σ-hole越大,使得形成的卤键作用更强。此外,结合位点原子所处的化学环境也会明显影响其成键强度,如CF3I与N2、NH3、N(CH3)3形成的C-I···N相互作用,卤键受体由N2变为NH3,卤键的强度增加了大约4倍;NH3中的氢原子被推电子基团-CH3取代后,卤键强度又进一步增加了约一倍,表明N原子的亲核能力进一步增强。由CF3I-H2O/H2S对比可知,随着卤键受体原子周期数的增加,卤键的强度变弱。CF3Cl、CF3I与各种小分子复合物的结构、卤键类型和卤键距离总结于表1,由于卤素原子的σ-hole位于其共价键的反方向,形成的卤键R-X···Y(Y为卤键受体)具有良好的方向性,所有R-X···Y的角度接近180°。
为了求出tmin,本文采用蒙特卡罗算法,生成k个随机整数xk,表示第k个时间段乘坐电梯的人数,然后根据电梯不用时所停放的层数、每一层人数的不同、电梯行驶每一层所消耗的时间、电梯最近接待原则等影响因素,考虑每个人等待电梯消耗的时间总和。
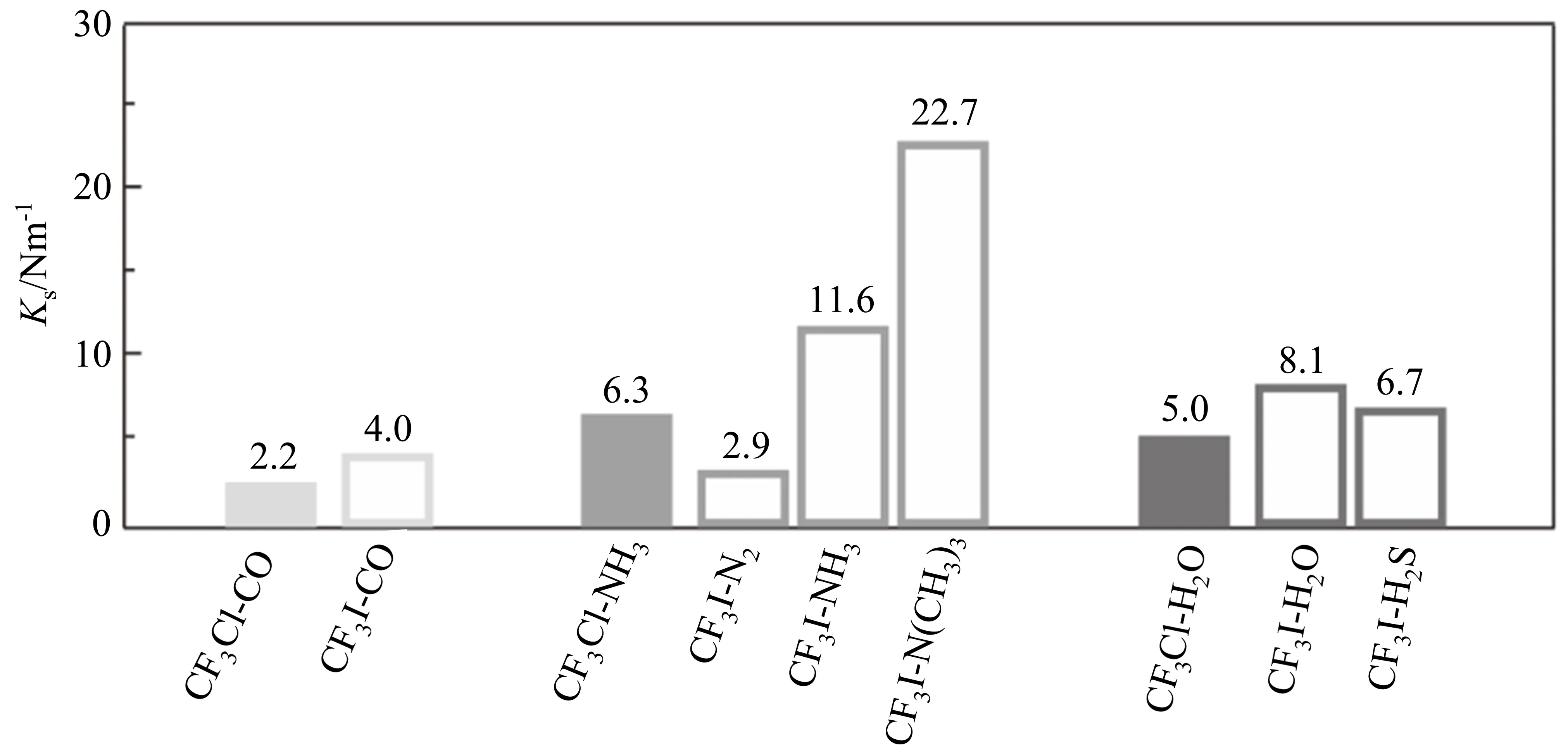
图5 CF3Cl、CF3I与不同族原子形成的卤键的伸缩力常数比较[94,95]Fig.5 Comparison of the stretching force constants in CF3Cl,CF3I complexes formed with different column atoms[94,95]
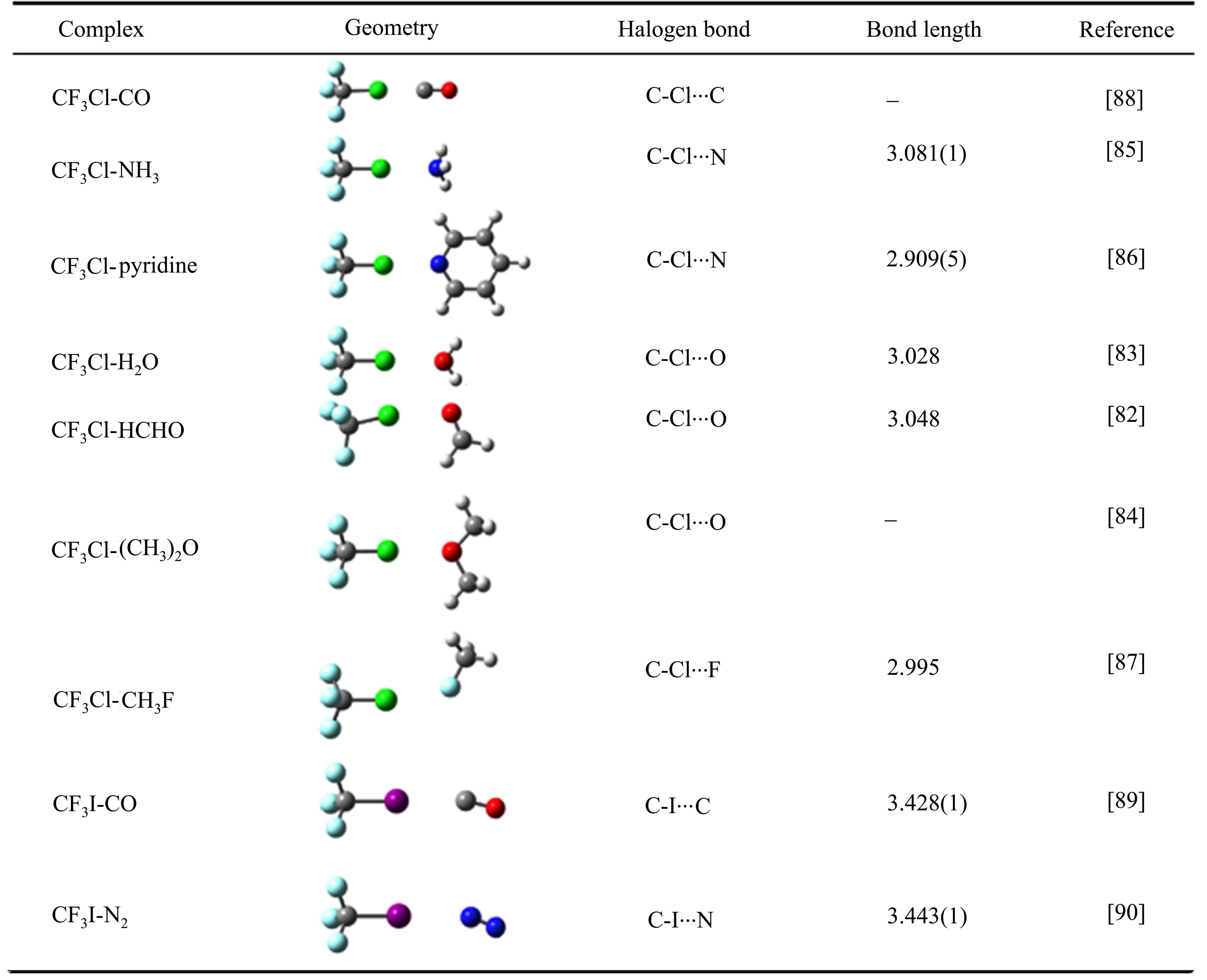
表1 CF3Cl、CF3I小分子复合物的结构、卤键类型和卤键键长Table 1 Geometry,type of halogen bond and bond length of the CF3Cl and CF3I complexes with small molecules
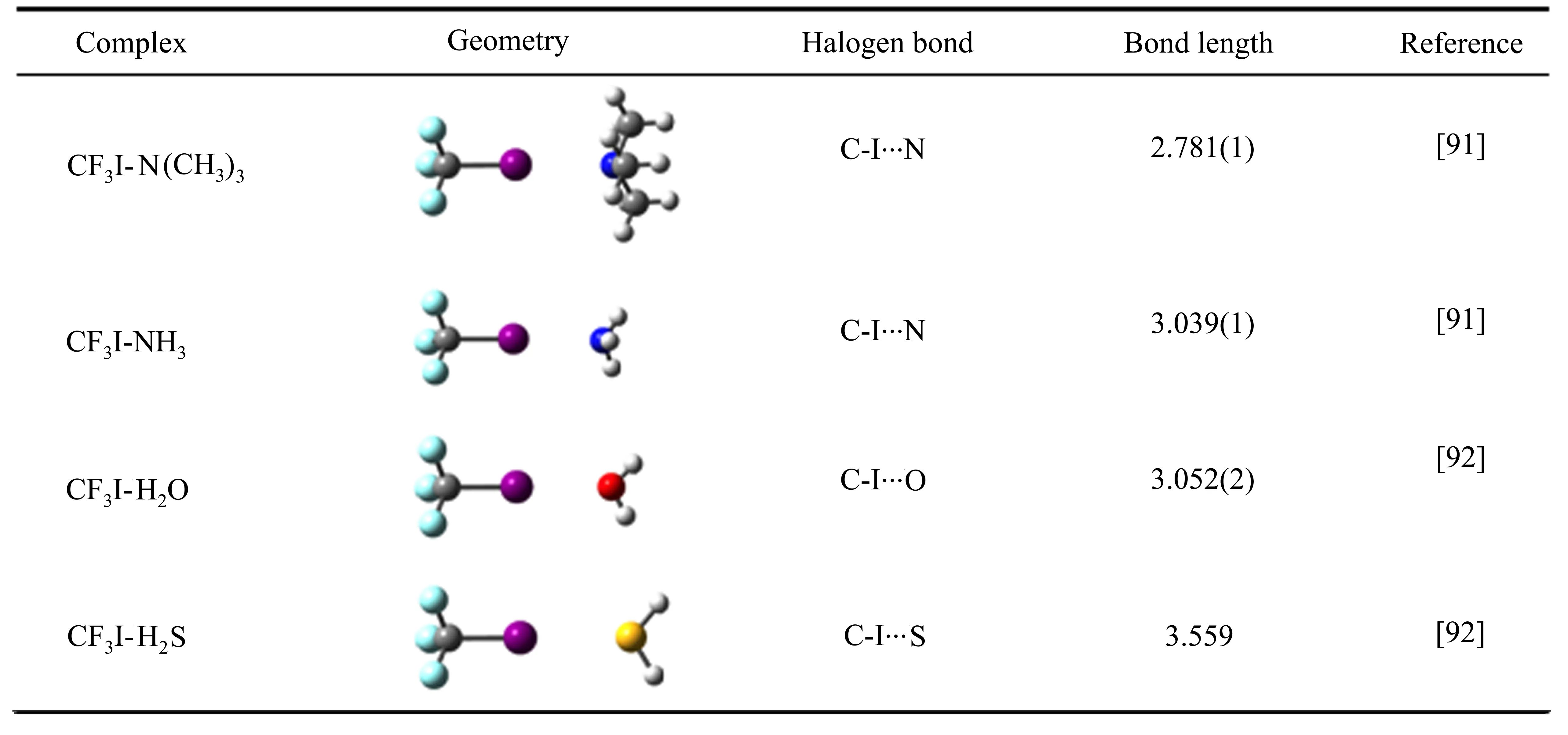
Continued
2.2 氧族σ-hole非共价相互作用
除了广泛研究的卤族元素的σ-hole相互作用,第VI主族的原子也能形成σ-hole,与富电子区域发生静电吸引,称为氧族σ-hole相互作用。目前,该族元素的转动光谱研究仅有以硫原子作为供体的文献报道。如在含硫的杂环分子四氟-1,3-二硫五氮杂环(C2S2F4)中,由于-CF2-的吸电子性,使得C2S2F4分子中的两个S原子上形成4个相同的σ-hole[96]。C2S2F4与CH2F2[97]、H2O[98]、NH3[99]复合物的转动光谱研究表明三个复合物分别以 S···F(0.29759(6)nm)、S···O(0.2912(5)nm)、S···N(0.2811(5)nm)作用为主导。在C2S2F4-H2O复合物中,水分子绕其C2轴的内转动(氢原子交换形成paraortho两个态)使每一个跃迁裂分为两条谱线,水分子内转动的势能面如图6所示。由于C2S2F4的对称性,HDO同位素异形体的低势垒转动同样会导致复合物产生两个等效的构型,但内转动会带来偶极矩方向的变化,从而可以从实验光谱解析获得两个态的能级裂分为21.342(5)GHz。
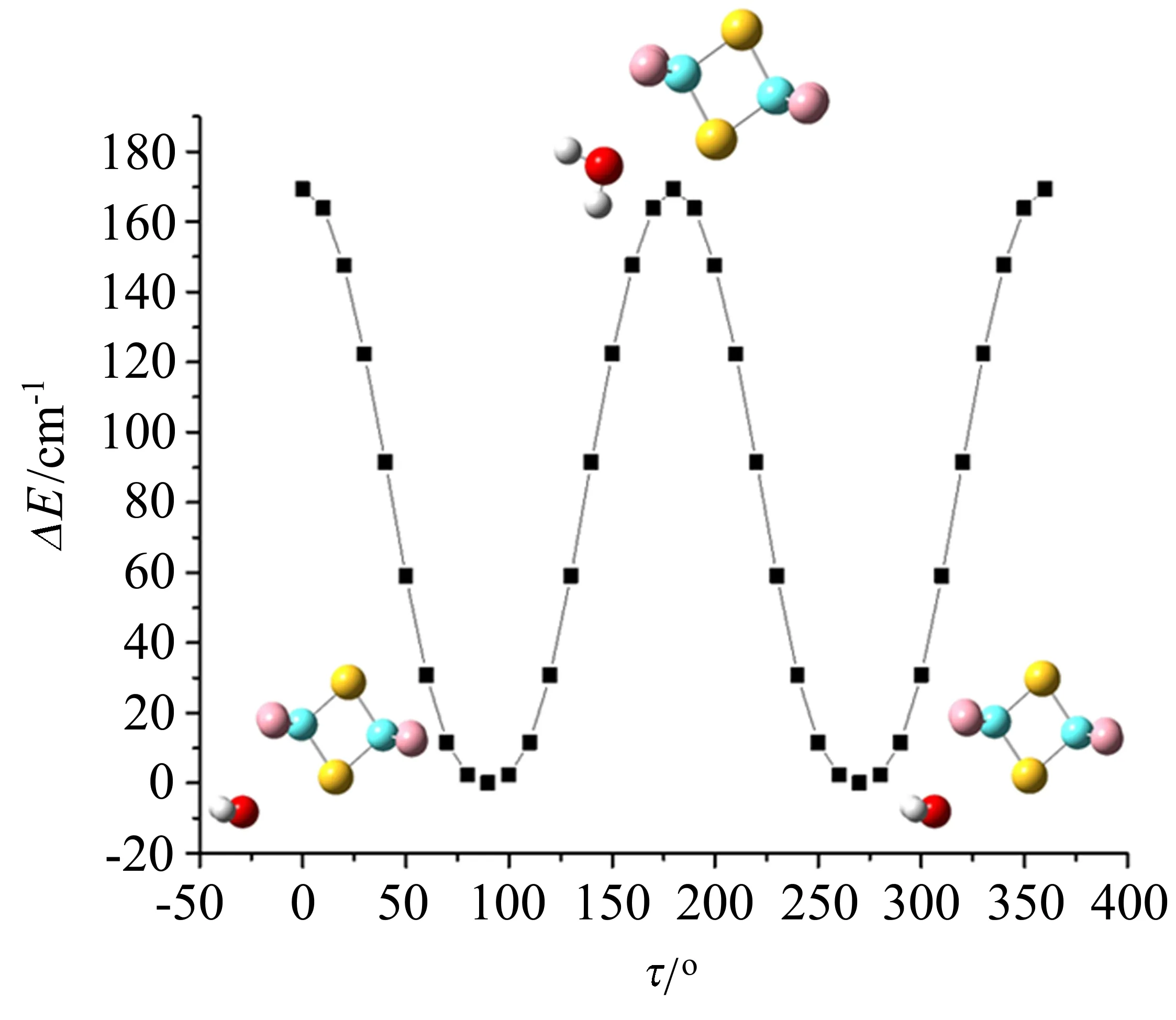
图6 MP2/6-311++G(d,p)理论水平计算的C2S2F4-H2O复合物中水绕其C2轴内转动的势能面[98]Fig.6 Potential energy surface for the internal rotation of water around its C2axis in C2S2F4-H2O complex calculated at the MP2/6-311++G(d,p)level of theory[98]
此外,由于F原子的强电负性,SF6分子中的S原子也可以形成σ-hole相互作用。如在SF6-NH3复合物中[100],NH3的N原子指向SF6中的S原子,形成N···S作用,NH3的三个H原子与SF6的三个F原子呈重叠式结构,如图7所示。

图7 SF6-NH3复合物最稳定构象的两个视角的结构[100]Fig.7 Structures with two views of the global minimum of SF6-NH3complex[100]
2.3 碳族σ-hole非共价相互作用
对于第IV主族的原子,从原子成键的角度而言,C、Si、Ge和Sn既可以形成σ键又能形成π键,使得IV主族的元素能够参与形成σ-hole和π-hole两种类型的非共价相互作用。自2013提出碳族相互作用的概念以来,已有很多关于碳族相互作用的理论计算和实验方面的研究,并且也报道了几篇相关的综述[13,101]。由于碳在许多生物和化学体系中具有非常重要的应用,如碳族相互作用可以稳定SN2反应的中间体[102]。因此,碳族相互作用的转动光谱研究主要集中在C原子的复合物(以下将C原子参与的碳族相互作用简称为碳键)。尽管C原子的范德华半径很小,但当它与强电负性的原子或基团成键时,在每个共价键的反方向可以形成一个低电子密度的区域σ-hole,如图8中CF4的分子表面静电势所示。因此,有些学者认为四氟甲烷(CF4)与电子供体如H2O[103,104]/环氧乙烷[105]、吡啶[106]等也是以C···O/N碳键结合[53]。在碳键被提出之前,转动光谱研究普遍认为两者形成了CF3···O/N卤键。但CF4中sp3杂化的碳原子形成四个等效的σ-hole,加上F原子的位阻效应,导致能够与富电位点作用的σ-hole区域非常有限,阻碍了其进一步的实际应用。最近气相转动光谱与X-射线单晶衍射研究表明,超分子合成子(supramolecular synthon)3,3-二甲基-四氰基环丙烷具有明显的sp3杂化σ-hole,能够与四氢呋喃的氧原子以静电力结合形成碳键,结合能约-11.2 kcal/mol[107],为进一步设计以sp3杂化σ-hole为中心的超分子化学提供了理论指导。
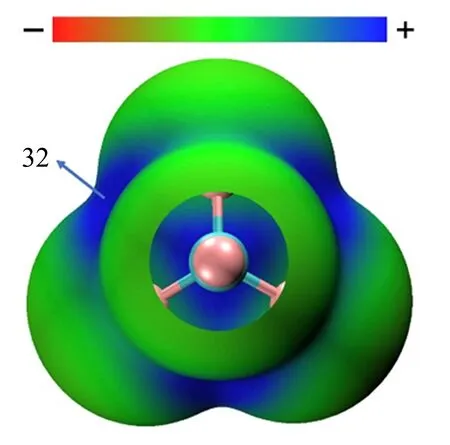
图8 CF4在MP/6-311++G(d,p)理论水平的分子表面静电势,能量单位为kcal·mol-1[13]Fig.8 Molecular electrostatic potential surfaces of CF4at the MP/6-311++G(d,p)level of theory.Energies in kcal mol-1[13]
3 π-hole非共价相互作用
在含有强电负性原子或基团的含π电子体系中,如果π电子离域到强电负性的原子或基团,在π电子体系上也会出现正电势或低电子密度区域π-hole,其具有部分空的π*轨道的特征,也容易与分子的富电子位点作用形成非共价键。
3.1 全卤代π-hole非共价相互作用
一氯三氟乙烯(CF2=CFCl)与H2O复合物[108]是首个转动光谱研究全卤代π-hole的非共价相互作用的实例。卤素原子取代乙烯中的氢原子,使分子中π电子被吸引到附近的卤素原子而在分子平面的两侧形成π-hole,图9(a)为CF2=CFCl-H2O复合物的分子间相互作用,可以发现水分子位于π电子体系的上方,且由于F原子的电负性大于Cl原子,所以水中的氧原子指向=CF2中的碳原子,形成孤对电子···π(lp···π)相互作用。类似地,NH3[109]和乙醚[110]也可以分别通过氮和氧的孤对电子与CF2=CFCl的π-hole作用形成lp···π非共价相互作用。
强电负性原子如F原子的取代也可以使芳香分子π电子被吸引到附近的卤素原子而形成π-hole。例如,六氟苯和五氟吡啶分别与H2O[111,113]和NH[3112]形成的四个复合物的转动光谱研究表明:H2O和NH3分子均与芳香环的π-hole相互作用。图9(b)、(c)中给出实验中观测到的六氟苯-水[111]和五氟吡啶-水[112]复合物构象的分子间非共价相互作用:水分子分别位于六氟苯和五氟吡啶环平面的上方,且水的O原子指向环中心,通过氧原子的孤对电子与芳香环的π-hole作用,形成lp···π非共价相互作用。两个复合物中水的内转动均使得谱线发生不对称裂分。水的内转动使两个H原子(费米子)发生等价交换,根据统计权重,两个态的谱线强度通常为1:3,其中强度较弱的态为基态。图10为五氟吡啶-水复合物的523←413跃迁的转动光谱,由于14N(核自旋量子数I=1.0)的核四极耦合效应,每个态的谱线进一步发生核四极裂分。
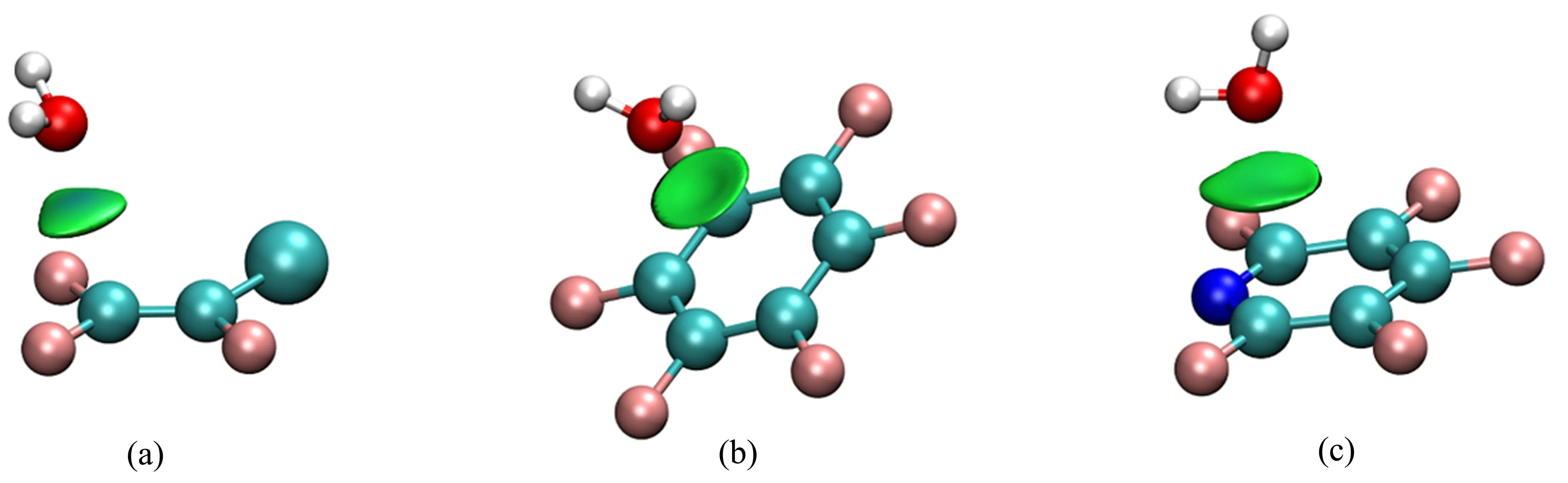
图9 一氯三氟乙烯-水(a)[108]、六氟苯-水(b)[111]和五氟吡啶-水(c)[112]复合物最稳定构象可视化的非共价相互作用Fig.9 The visualizations of the non-covalent interactions occurred in the observed isomer of the CF2=CFCl-H2O(a)[108],C6H6-H2O(b)[111]and pentafluoropyridine-H2O(c)[112]complexes
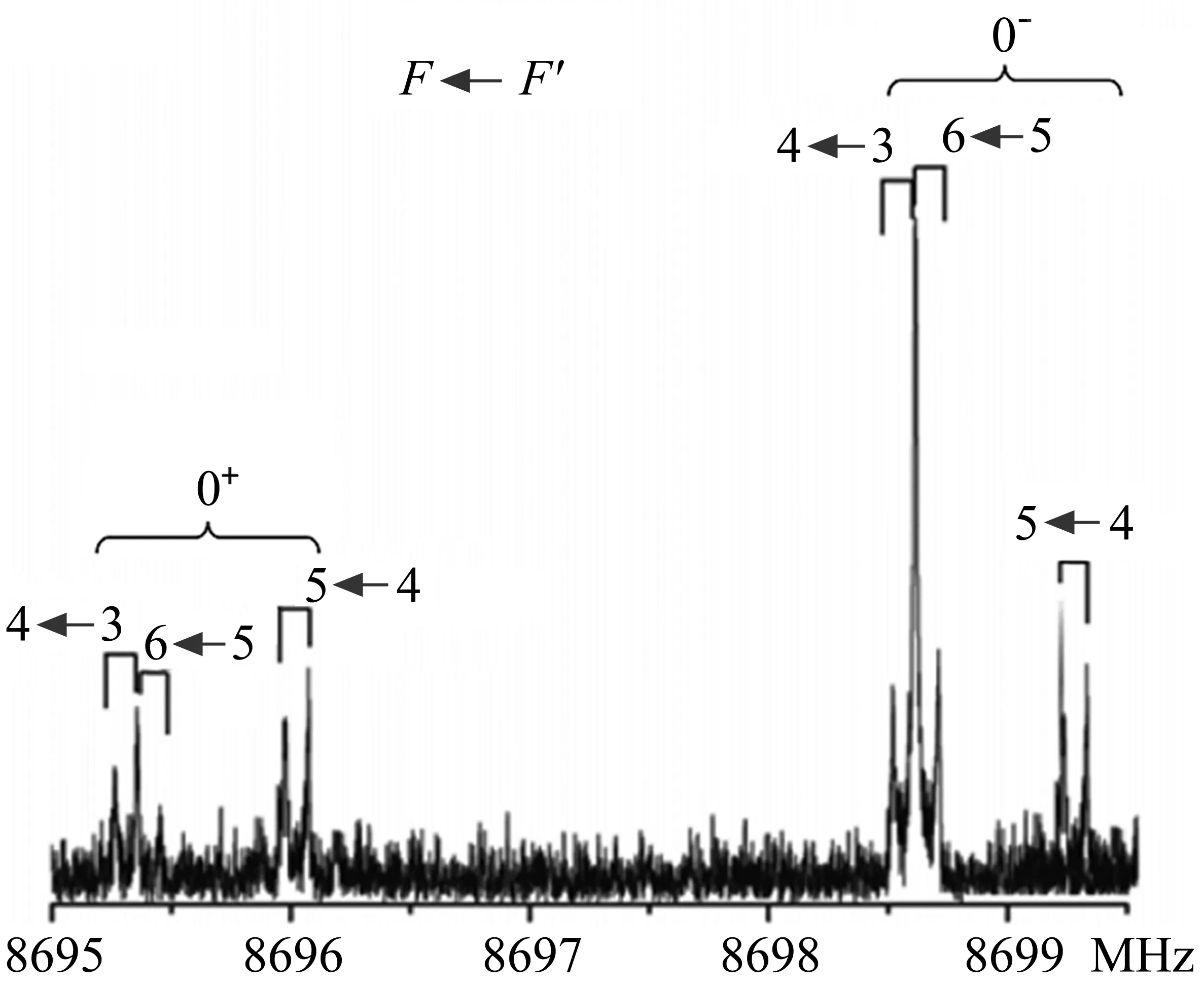
图10 五氟吡啶-水复合物523←413跃迁的转动光谱图,每条谱线均显示出多普勒裂分(⊓)[112]Fig.10 Rotational spectrum of the 523←413transition of the pentafluoropyridine-water complex,each component line exhibits an instrumental Doppler doubling(⊓)[112]
最近,苯甲醛-CF4复合物[5]的转动光谱实验观测到的结构是CF4位于芳香环上方。根据转动跃迁谱线裂分的解析,可以确定-CF3绕着C15-F16键发生内转动(见图11),这可以直接指出该复合物以F···π非共价相互作用为主导,即F16原子被锚定在π上方。但是,F原子与π体系往往被认为均是电子供体,它们之间的相互作用似乎违背了化学直觉。主相互作用轨道分析(PIO)[114]表明这种独特的F···π非共价相互作用通过F孤对电子与苯甲醛π-hole相连。同时PIO分析也指明,在苯-CF4复合物中也存在同样的F···π非共价相互作用。因此,得出以下猜测:1)全卤代可能并不是芳香环π-hole特性表现出来的必要条件;2)类似于CF4的电负性很大的配体分子也能诱导芳香环的π-hole特性;当然这一点需要更多的实验案例验证。
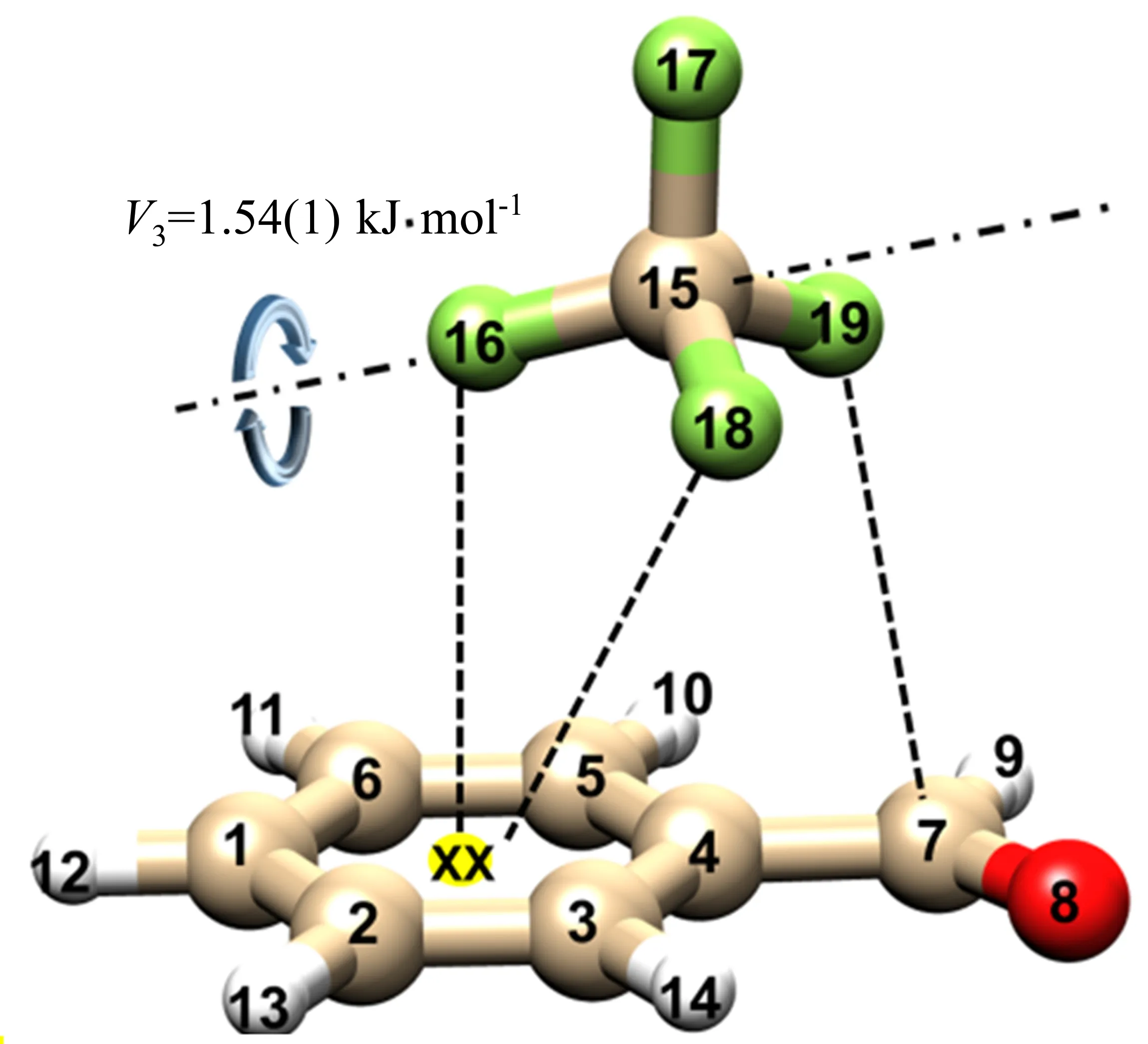
图11 观察到的苯甲醛-CF4构象的结构、原子编号,芳香环中心的XX为虚原子[5]Fig.11 Shape of the observed isomer of benzaldehyde-CF4with atomic labelling.A “dummy atom”labelled“XX”is placed in the centre of the aromatic ring[5]
3.2 sp杂化π-hole非共价相互作用
碳原子可以采用多种杂化轨道参与成键,对于采取sp杂化的CO2,由于氧原子的电负性大于碳原子,使得碳原子周围形成低电子的密度区域π-hole,易与分子的富电子位点作用形成sp杂化的π-hole非共价相互作用[115]。现在已经有很多CO2参与形成复合物的转动光谱研究,表明CO2与不同的配体分子作用,两者间的非共价相互作用方式可能不同。比如,CO2与HF[116]或HCl[117]通过C=O···H氢键作用,形成几乎线性的结构,而与HBr[118]则以C···Br碳键结合形成T形结构。分子与CO2之间同时存在氢键和碳键时,以何种非共价键相互作用为主导则与电子供体的分子环境密切相关。如表2所示,通过对转动光谱观测到复合物的构象进行键径分析及键能计算表明,甲酰胺-CO2[119]和甲醛-CO2[120]两个复合物主要以C···O碳键结合,而在甲酸-CO2[121]中,O-H···O氢键则为主导的非共价键相互作用模式。从表2还可以发现,实验中观察到了甲酰胺与CO2形成的两个稳定构象,CO2中的碳原子与羰基氧相互作用,而CO2的一个氧原子既可以与氨基的氢作用,也可以与羰基碳上的氢作用。通过选择几条构象I和构象II的特定谱线,利用公式[122]

表2 甲酰胺-CO2、甲醛-CO2以及甲酸-CO2构象的QTAIM[123](Quantum theory of atoms-in-molecules)分析及键能Table 2 QTAIM analyses and bonding energies of formamide-CO2,formaldehyde-CO2and formic acid-CO2complexes

可以计算得到两个构象在分子束中的比例约为NI/NII=16:1。其中,I为测定谱线的峰高、μ为电偶极矩、γ为预测的谱线强度、ν为转动跃迁的频率值。
非常有趣的是,虽然CF3Cl是研究卤键的典型模型分子,如前所述,其与H2O[83]、CH2O[82]、NH3[85]、CO[88]、CH3F[87]、CH3OCH3[84]以及吡啶[86]等均是与Cl原子的正电势区域即σ-hole作用形成卤键。然而,在CF3Cl分子中,除了F吸电子效应引起的Cl σ-hole外,Cl原子周围还存在一个孤对电子“赤道带”[124],如图12所示。当CO2与CF3Cl形成复合物时,Cl赤道带被CO2中的π-hole“激活”而形成C···Cl碳键。该转动光谱研究也表明,弱的CO2π-hole更倾向于与弱的Cl赤道带电子供体结合,而非F孤对电子,为揭示非共价相互作用的本质提供了新思路。
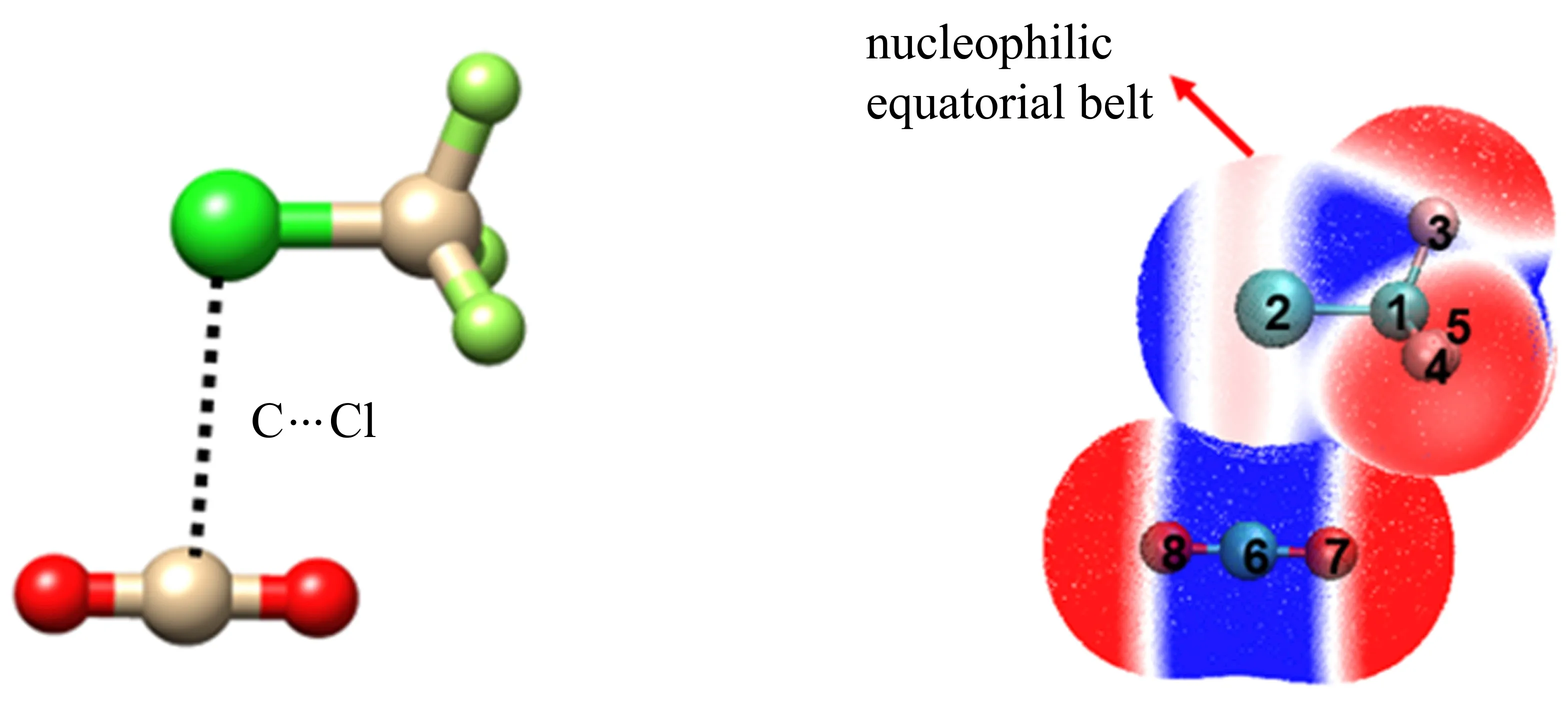
图12 实验观察到的CF3Cl-CO2复合物的结构和分子间的非共价相互作用[124]Fig.12 Observed structure of CF3Cl-CO2and its intermolecular noncovalent interaction[124]
氮族π-hole相互作用是第V主族原子的π-hole与其它分子的亲核位点之间的一种相互作用[101]。在提出氮族π-hole相互作用这个概念以前,转动光谱已经研究了采用sp杂化的N2O与一些小分子复合物之间形成的氮族π-hole相互作用,例如,转动光谱的研究结果表明N2O与CO[125]、HC≡CH[126]、HCl[127]、HF[128]、H2O[129]和 NH3[130]分别通过 N···C、N···π、N···Cl、N···F、N···O 和 N···N 键相互作用,并且除了N2O-HC≡CH以外,其余复合物的结构均为T构型。这种氮族π-hole相互作用是否适用于更大尺寸的分子体系,需要找到更多合适的模型分子进行转动光谱研究。
3.3 sp2杂化π-hole非共价相互作用
对于sp2杂化碳原子形成的π-hole非共价相互作用,转动光谱研究主要集中在含有羰基的醛和酮类化合物,如甲醛[131]、乙醛[132]与吡啶复合物的转动光谱研究结果表明两个复合物中均以醛的羰基作为碳键供体与吡啶的N原子相互作用形成C···N碳键。当形成复合物的两个配体分子均含有羰基时,作为碳键供体的羰基存在竞争。例如甲醛与苯甲醛形成的复合物,转动光谱研究证实甲醛的羰基作为碳键供体,而苯甲醛中的氧原子提供碳键受体,形成C···O碳键[133]。但是,转动光谱研究表明3-氧杂环丁酮(3OXT)的羰基却显示了比甲醛更强的碳键供体能力,并与甲醛的氧原子相互作用形成C···O碳键[134]。在该体系中,还完成了13C和两个羰基的18O同位素异形体的转动光谱研究并得到每个单同位素异形体的实验光谱参数,得到同位素的转动光谱图如图13所示。基于测定的同位素异形体的转动常数,结合Kraitchman方程[135]

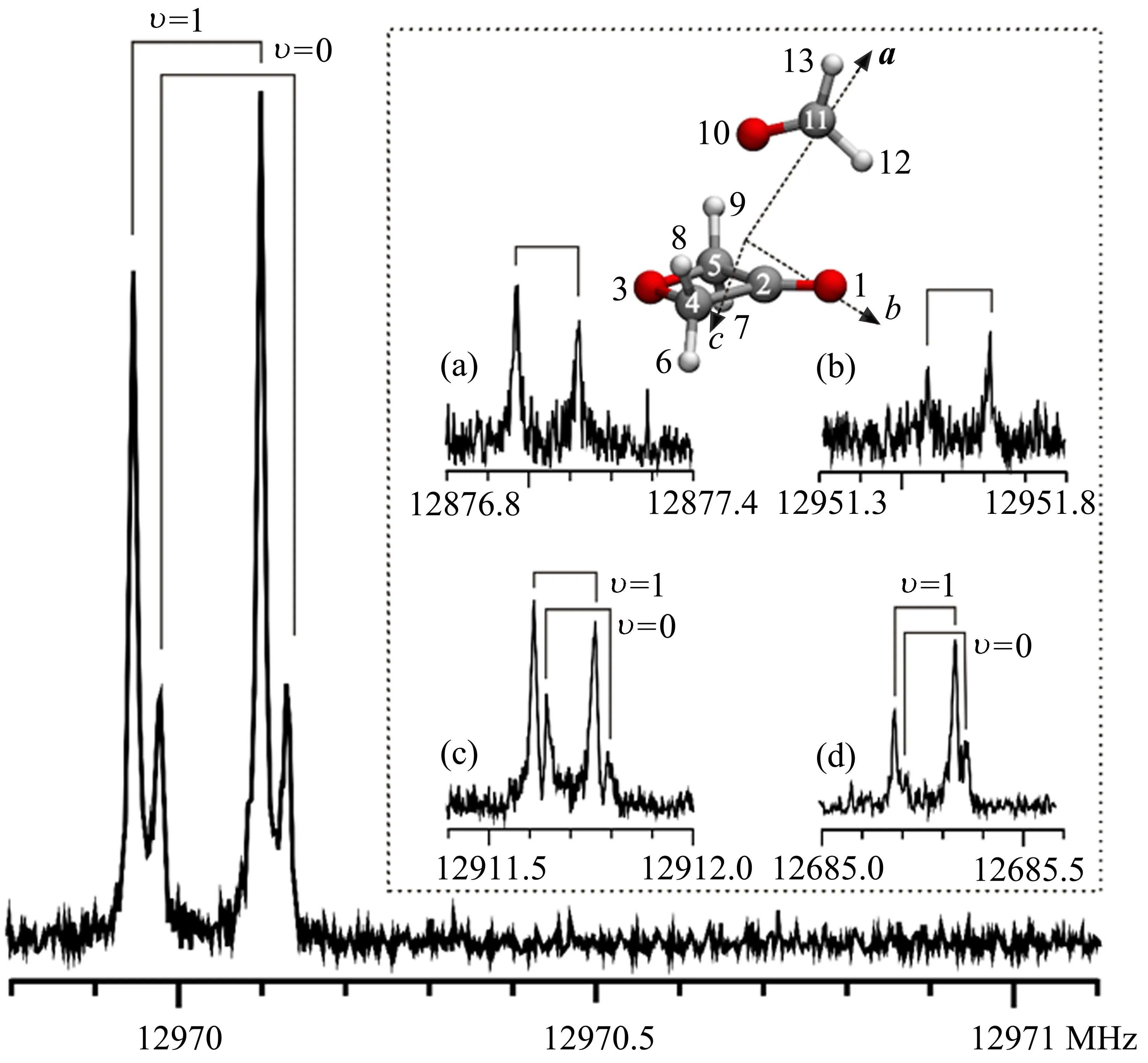
图13 3OXT-H2CO复合物的母体、13C4/5(a)、13C2(b)、18O3OXT(c)和18OH2CO(d)同位素异形体41,3←31,2跃迁的转动光谱(循环次数:1024)。由于甲醛的内转动18O3OXT和18OH2CO同位素异形体的转动跃迁出现了清晰的裂分,而由于较差的信噪比没有观察到13C同位素异形体的裂分。每条谱线均显示出多普勒裂分(⊓)[134]Fig.13 Rotational spectrum of(cycles=1024)41,3←31,2transition of the parent species,13C4/5(a),13C2(b),18O3OXT(c)and 18OH2CO(d)isotopologues of 3OXT-H2CO complex.The rotational transitions of the18O3OXTand18OH2CO isotopologues exhibit clear splitting due to the internal rotation of H2CO,while poorer S/N inhibits its visibility of the internal rotation splitting in the 13C species.Each component exhibits the instrumental Doppler doubling(⊓)[134]
即可计算得到每个单取代原子的坐标,进而准确推导出复合物的实验结构。公式中,Pgg(g=a,b或c)为平面矩,如M为取代前分子或复合物的质量,Δm为同位素取代后的质量变化。3-氧杂环丁酮的同分异构体β-丙内酯与两个和三个水的复合物中[136],羰基与水的氧原子之间也存在C···O碳键。对于更大尺寸的二苯甲酮(Ph2CO),它的一水合物和二水合物均是通过 O-H···O 和 C-H···O 氢键作用,但其与三个水分子的团簇中,C···O 碳键和 O-H···O、O-H···π氢键网共同稳定了复合物[137],存在碳键作用的三水合物的结构如图14所示。对于这种分子团簇的转动光谱研究,CP-FTMW光谱仪通常会更具有优势,图15为CP-FTMW光谱仪实验记录的二苯甲酮水合物的宽带转动光谱图。

图14 吡啶-甲醛[131]、3-氧杂环丁酮-甲醛[134]和二苯甲酮-(H2O)3[137]团簇中羰基形成π-hole相互作用的自然键轨道重叠图Fig.14 Overlap of the natural bond orbital of carbonyl π-hole interaction in pyridine-formaldehyde[131],3-oxetanone-formaldehyde[134]and benzophenone-(H2O)3[137]clusters

图15 二苯甲酮与水的宽带转动光谱。上方黑色的线表示实验光谱(5.6×106个自由感应衰减信号,22 h采集时间)。光谱图中去除了二苯甲酮单体分子及其同位素异形体的所有跃迁。下方的线为用实验光谱参数、转动温度为1 K和理论偶极矩预(μa/μb/μc=1.1/1.4/0.1)测得到的光谱:Ph2COH2O、Ph2CO-(H2O)2和Ph2CO-(H2O)3的谱线分别为红色、蓝色和绿色。下面的三个图为三个复合物代表性的转动跃迁[137]Fig.15 Broadband rotational spectrum of Ph2CO with water.The black upper trace corresponds to the experiment(average of 5.6 million FIDs,22 h of acquisition time).All rotational transitions arising from the Ph2CO monomer and its isotopologues have been removed.The lower traces represent simulations produced with the experimental spectroscopic parameters,a rotational temperature of 1 K,and the theoretical dipole moment components(μa/μb/μc=1.1/1.4/0.1)for Ph2COH2O(red),Ph2CO-(H2O)2(blue),and Ph2CO-(H2O)3(green),respectively.At the bottom,parts of the spectra highlight representative transitions from each of the three complexes[137]
关于氮族原子采用sp2杂化形成π-hole相互作用的转动光谱研究相对较少,目前仅有硝基乙烷-三甲基胺复合物[138]的报道,观察到的最稳定构象的结构如图16所示。实验确定了N···N相互作用的距离在0.3044~0.3081 nm之间,并且发现其结合能与相对强的中性氢键具有相同的数量级。不同的量子化学计算也表明N···N相互作用要强于体系中的C-H···O和C-H···N氢键。
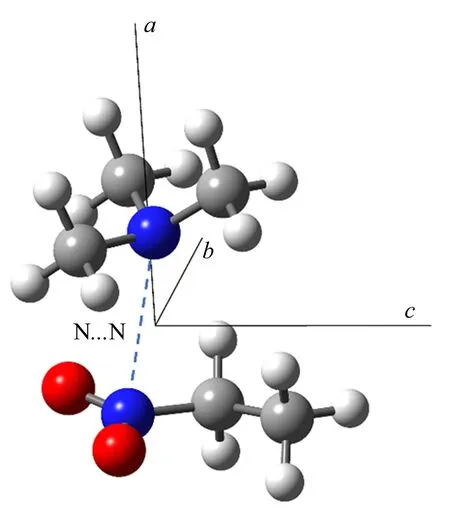
图16 实验观察到的硝基乙烷-三甲基胺复合物的结构[133]Fig.16 Observed structure of the complex of nitroethane-trimethylamine[133]
转动光谱研究不等性sp2杂化的SO2与小分子形成的复合物,也观察到了π-hole的非共价相互作用,即SO2的硫原子可以参与形成氧族π-hole非共价相互作用。在IUPAC定义[139]氧族相互作用之前,上世纪90年代便有很多SO2复合物的报道,如SO2与H2O[140]/甲醇[141]、N(NH3)3[142]、CO[143]形成的复合物,它们实际上分别是通过S···O、S···N、S···C硫键相互作用。在SO2与含π分子形成的复合物中,如乙炔[144]、丙炔[145]、丙烯[146]、丁二烯[147]、苯[148]、甲苯[149]和呋喃[150],也发现了S原子位于π电子上方的结构,形成S···π相互作用。最近,转动光谱研究发现硫原子不仅可以作为氧族相互作用的供体,还能作为受体,在SO2与(CH3)2S形成的复合物[151]中最大的分子间相互作用为S···S相互作用,复合物以SO2中的硫原子作为亲电的π-hole与(CH3)2S中硫原子的孤对电子相互作用,实验推导出S···S间的距离为0.2947(3)nm,远小于两个硫原子的范德华半径之和(0.360 nm)[152]。SO3的硫原子采取等性sp2杂化,也可以形成π-hole非共价相互作用。比如SO3-SO2复合物[153],两个硫原子均具有π-hole的正电势区域,由于SO3含有更多的强电负性氧原子,所以SO3中的硫原子提供更强的π-hole与SO2的氧原子形成S···O相互作用。此外,SO3同样能够提供氧族π-hole与吡啶[154]、氰类化合物[155,156]和三甲胺[157]作用形成S···N相互作用。
4 总结和展望
简述了转动光谱的基本原理、发展历史以及研究非共价相互作用的优势,总结了转动光谱在研究σ-hole和π-hole非共价相互作用方面的最新进展。实验手段及量子化学理论方法的快速发展使研究者对非共价相互作用力的认识有了长足进步。与共价键完全不同的是,较弱的非共价相互作用使分子间存在多种相等或者相近能量的结构形式,其受到分子本身化学环境及配体分子的影响明显,这也为追溯其物理本源和阐释更加普适的规律提出了挑战。在分子团簇中,其结构稳定性往往由多种非共价相互作用共同支配,而且随着团簇尺寸的变化,不同形式的相互作用参与比重也发生着变化,这些不确定性大大增加了对其研究分析的难度。从研究对象上来看,除了建立可靠的分子模型以及从分子水平对不同的新型非共价相互作用进行表征外,如何从这种小分子模型拓展到对超分子化学、晶体工程、新材料新药物设计等更有实际参考价值的大分子体系上则需要对各类相互作用进行更系统和更深入的研究。
虽然转动光谱对分子体系的质量分布非常敏感,是从结构上和能量上研究非共价相互作用的有效手段,但受到必须在气相中进行的限制,为在线制备一些难以气化或特殊的分子复合物(如预反应复合物)提出了很大的挑战。将超声射流冷却结合激光烧蚀[158]、直流放电[159]、快速混合[160]等在线制备技术应用于高分辨分子光谱研究可能是有效的解决手段。但在线制备体系的转动光谱非常复杂,往往不能制备纯目标分子体系,因此采集到的转动光谱是前体分子单体、复合物及各种碎片等的跃迁谱线相互交错叠加而成,大大增加了光谱解析难度,这就更加需要高精度的量子化学计算作为指导,以及未来利用机器学习等智能分析手段来提高光谱解析效率。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
西华大学学报(自然科学版)(2021年3期)2021-05-17
西华大学学报(自然科学版)(2020年6期)2020-10-15
电子制作(2018年10期)2018-08-04
红领巾·探索(2018年12期)2018-01-26
电子制作(2017年24期)2017-02-02
药学研究(2015年11期)2015-12-19
