
基于转录组测序筛选和田羊毛囊周期性变化过程中差异表达miRNAs以及初步分析
2021-12-11 12:09阿布来提苏来曼李志强侯晨曦决肯阿尼瓦什依明苏来曼
草食家畜 2021年6期
孙 爽,阿布来提·苏来曼,李 彬,李志强,侯晨曦,决肯·阿尼瓦什,依明·苏来曼*
(1.新疆农业大学动物科学学院,乌鲁木齐830052;2.新疆畜牧科学院畜牧研究所,乌鲁木齐830011)
和田羊属于短脂尾异质半粗毛羊,具有耐干旱炎热、耐粗饲、抗病性强等优点[1,2],其毛色洁白,富有光泽,纹长,纤维弹性强,是织制地毯和提花毛毯的优质原料[3,4]。现有的和田羊经过多年的保种选育仍存在体型较小、产毛量低、底绒细少、干死毛多等缺陷[5]。研究表明,羊毛的品质与产量主要由毛囊决定[6],而后者存在周期性生长的特点,一个生长周期包括生长期、退行期和休止期[7]。对于牛、羊哺乳动物来说,生长期一般在每年的4-11月;退行期一般是在每年的12月到次年的1月[8],休止期一般在每年的2-3月,哺乳动物皮肤毛囊的生长周期主要是由外界温度变化诱导,一般生长周期与每年的季节交替保持一致。王丽[9]研究发现和田羊毛囊形态结构在2月以后迅速变化,2-8月,和田羊总毛囊密度整体呈增长趋势,处于生长期,2月时毛囊活性较弱,4-8月毛囊群丰富,生长密集,活性较好,为和田羊毛纤维的生长发育提供良好的生理基础。山区型和田羊毛囊密度在6月到8月增长趋势尤为显著,因山区型和田羊生活在海拔1 900 m左右的山腰上,气温较低且温差大,结合本课题组的长期观察研究,山区型和田羊的毛囊生长期一般在5月,退行期一般在10月,休止期一般在1月。
毛囊周期性生长发育与许多代谢过程有关,毛囊在特定的细胞中,可以选择性的表达或抑制某些基因,在这过程中有大量调控因子参与其中,如miRNA、LncRNA等,涉及多种复杂的调控通路,如Wnt、Notch、TGF-β、Hedgehog、MAPK、TNF等通路,还有大量细胞参与其中,相互协调,共同完成了毛囊生长发育过程[10]。miRNAs是一类机体自身基因转录产生的、长度约为18~25个核苷酸的内源性小分子非编码RNA,能通过反向互补配对的方式与mRNA的3'UTR靶向结合,进而在转录后水平影响mRNA的稳定性或蛋白翻译过程[11]。研究表明,miRNAs在毛囊发育过程中发挥着重要的调控作用。Zhang[12]等利用芯片和生物信息学技术比较内蒙古绒山羊和美利奴绵羊的耳尖和体侧区皮肤中的差异表达miRNAs,发现有19个miRNAs在体侧皮肤区特异表达,15个miRNAs可能在毛囊发育中上调。曲海娥[13]发现miRNA-125b在细毛羊和绒山羊中作用于FGFR2和MXD4这两个靶基因然后调控毛囊的生长发育。李勤群[14]发现miRNA-148b和miRNA-320在藏绵羊毛囊发育生长期的表达量显著增加。唐晓慧[15]通过对毛囊周期性变化中3个时期转录组测序的分析,在毛囊生长期、退行期、休止期发现差异表达miRNAs分别有10、27、7个,并同时发现miRNA-184、miRNA-205的靶基因富集在Wnt通路中。
研究和田羊毛囊周期性的差异表达miRNA,发掘绵羊miRNA图谱,探索miRNA在周期表达的变化,再通过靶基因预测及功能富集分析,为进一步研究其在绵羊周期调控过程中的功能提供帮助,为筛选和田羊毛性状关键基因提供前期基础,最终为在实际育种生产中通过调控miRNA及其靶基因的表达,人工干预绵羊羊毛周期生长发育实现增产提供科学依据。目前和田羊毛囊周期性变化中差异表达miRNAs的筛选及相关分析对和田羊毛生长发育和高品质地毯毛形成的分子机制方面的研究还未见报道。
本研究利用RNA测序(RNA-sequencing)技术鉴定了和田羊毛囊周期性变化过程中不同时期毛囊的差异表达miRNAs,并通过生物信息学分析鉴定了差异表达miRNAs的潜在生物学功能,结果表明miRNAs可能参与调控和田羊毛囊周期性变化,为进一步探究miRNAs对和田羊毛囊周期性变化中的调控机制提供理论基础,同时为和田羊品种的选育提供了一定的科学依据。
1 材料与方法
1.1 试验材料
1.1.1 试验动物
在新疆和田地区于田县昆仑种羊场选择3只周岁、健康无病、体况均一的山区型和田母羊作为试验对象。经过统一饲养管理,分别于毛囊生长的生长期(5月份)、退行期(10月份)和休止期(次年1月份)采集数块同一侧肩胛部位大小约1 cm2的皮肤组织,分三份置于液氮中保存备用。
1.1.2 试剂及仪器
Infinite M200酶标仪,DYY-6C型电泳仪,7900荧光定量PCR仪,Gel Doc 2000型凝胶成像仪,反转录试剂盒(诺唯赞公司),Trizol试剂(Invitrogen公司),荧光定量PCR试剂盒(Tiangen公司)。
1.2 试验方法
1.2.1 提取皮肤组织RNA将三个时期的皮肤组织样分别在加入液氮的灭菌研钵中研磨至粉末状,用Trizol法提取RNA,检测提取RNA的浓度与质量。合格的RNA置于-80℃保存,备用。本试验所有操作均在低温下进行.
1.2.2 cDNA的制备和文库的构建
cDNA制备:于灭菌的eppendorf管内依次加入2X miRNA RT Reaction、10 μL Buffer、2 μL miRNA RT Mix、500 ng RNA,最后加RNase-free water配成20 μL的体系,按42℃、60 min;95℃、3 min程序进行反转录,产物置于冰箱-20℃保存。
文库的构建:由上海鲸舟基因科技有限公司完成。
1.2.3 转录组的高通量测序及分析
(1)由上海鲸舟基因科技有限公司采用illumina Xten测序平台的双端150 bp测序模式对样本进行高通量测序,共构建3个时期9个转录组测序文库。
(2)对测序原始数据进行质量评估,并利用Seqtk软件过滤原始数据(raw reads)得到高质量数据(clean reads)。
(3)将clean reads分别比对到miRBase、Genome数据库羊的基因组(Oar-v3.1.91)上,确定已知的miRNAs,将未知的smallRNA与Rfam非编码数据库进行比对,对小RNA片段进行分类注释和数量统计,再利用miRDeep软件对未知的小分子rRNA根据miRNAs前体的标志性发夹结构来预测新的miRNAs。
(4)以下将以P1、P2、P3,Ⅰ、Ⅱ、Ⅲ分别代表生长期、退行期、休止期,P1、P2、P3为同一时期3(如01180635Ⅰ、00617149Ⅰ、01180656Ⅰ)个样本的平均值,分三组对比组对比,P2/P1对比组,P2为处理组,P1为参考组;P3/P1对比组,P3为处理组,P1为参考组;P3/P2对比组,P3处理组,P2参考组。将每个样本的reads比对到miRBase上,计算miRNAs表达量(count数)。miRNAs表达量的筛选采用CPM计算度量指标(counts per million),再利用DESeq软件对样本处理组与样本参考组进行差异表达分析,在两两对比比较组选择Pvalue≤0.05并且log2(foldchange)≥1的miRNAs作为差异表达miRNAs。
1.2.4 差异表达miRNAs靶基因的预测
利用miRanda、miRDB、TargetScan等工具对差异表达miRNAs进行靶基因预测,取预测结果的交集作为预测的最终结果。
1.2.5 靶基因的GO和KEGG富集分析
将预测的靶基因分别利用GO seqR和KOBAS进行GO和KEGG功能富集来分析靶基因的功能及信号通路以研究差异表达miRNAs潜在的功能。
1.2.6 qRT-PCR
为了验证转录组测序的准确性,利用qRT-PCR技术检测了P3与P1两个时期差异表达较大、CPM值较高的7个miRNAs进行相对表达量分析。(1)根据7个miRNAs的成熟序列设计qRT-PCR的上游引物,下游引物由试剂盒(miRcute Plus miRNA qPCR Kit(Tiangen FP411)提供,引物序列见表1,由上海生工生物工程技术服务有限公司合成。(2)以1.2.2获得的cDNA为模板,以U6为内参基因,以SYBR Green染料法进行7个miRNAs相对表达水平的实时定量分析。qRT-PCR反应总体系为10 μL:Mix(2X)5 μL、Amplification primer 1(2 μM)1 μL、Reverse Primer(2 μM)1 μL、cDNA 1 μL、ddH2O 2 μL,扩增程序为50℃2min;95℃,10 min;95℃15 s,60℃1 min,40个循环;95℃,15 s,60℃,15 s,95℃,15 s,每个样品设置3个重复。结果以2-ΔΔCt方法进行目的基因相对表达量的计算,其中ΔCt=Ct目的基因-Ct内参基因,以01180635Ⅰ作为对照组,ΔΔCt=ΔCt实验组-ΔCt对照组。与测序结果进行对比,用excel作图表示。

表1 引物序列参数
2 结果与分析
2.1 提取RNA检测
提取的质量检测结果如表2,OD260/OD280在1.8~2.1之间,浓度均在50 ng/μL以上,RIN在7.0以上,表示所提RNA浓度高且无蛋白质、DNA等污染,符合后期反转录的要求,可用于后续的实验。
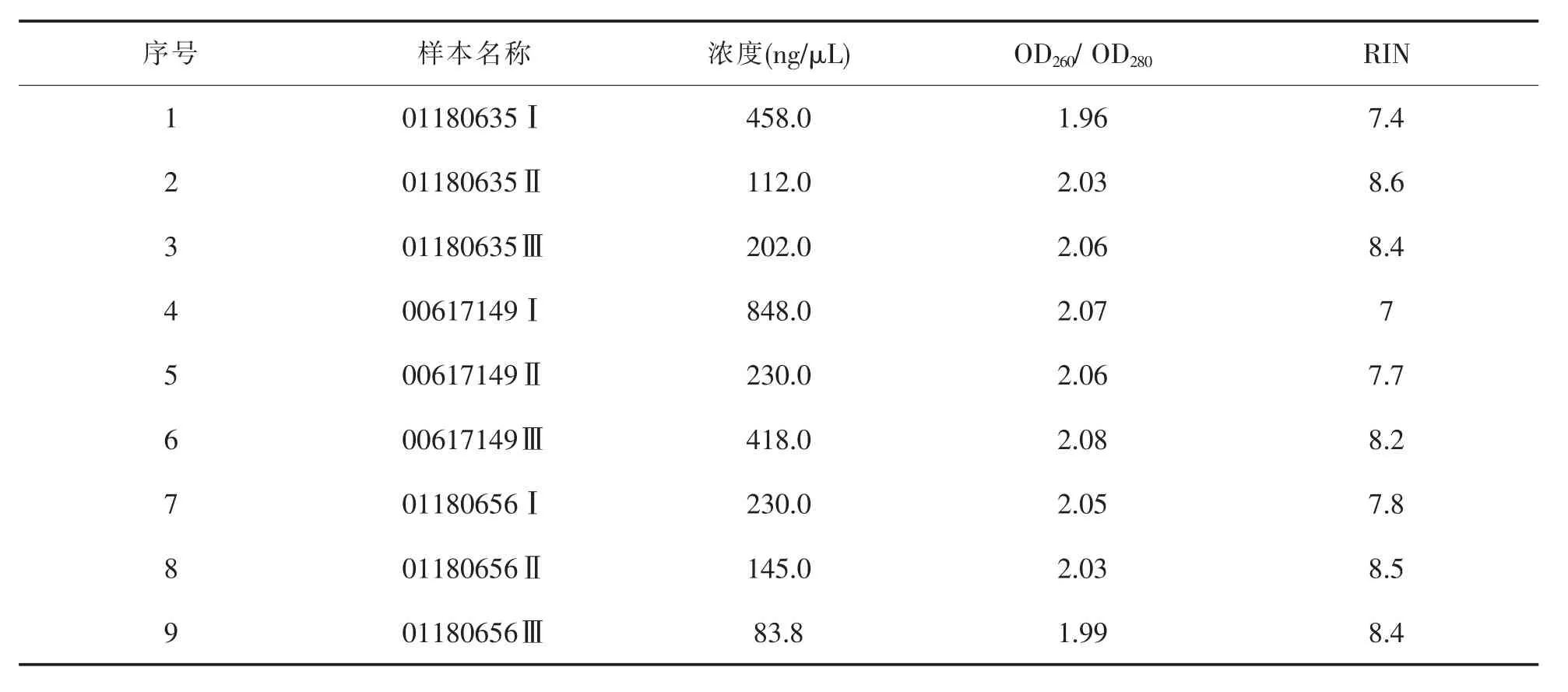
表2 提取RNA质量检测结果
2.2 转录组测序及分析
2.2.1 测序质量与数据处理情况
平均每个样品测序获得约24 M的数据量,平均每个样本获得24 768 122个原始数据,处理后平均每个样本获得21 499 205个clean reads,过滤各项有影响的reads得到的结果见表3,高质量数据占比较高为86.80%,30%在所有样本中均为90%以上,说明单个碱基的错误率非常低,表明产出的reads质量可靠;9个文库所得序列长度大部分位于19~25 nt之间,其中长度为22 nt的序列所占比例最大,符合miRNAs长度分布特征。图1为样本S0617149I的reads长度占比情况。以上均表明测序数据质量较好,符合试验要求,可用于后续结果分析。
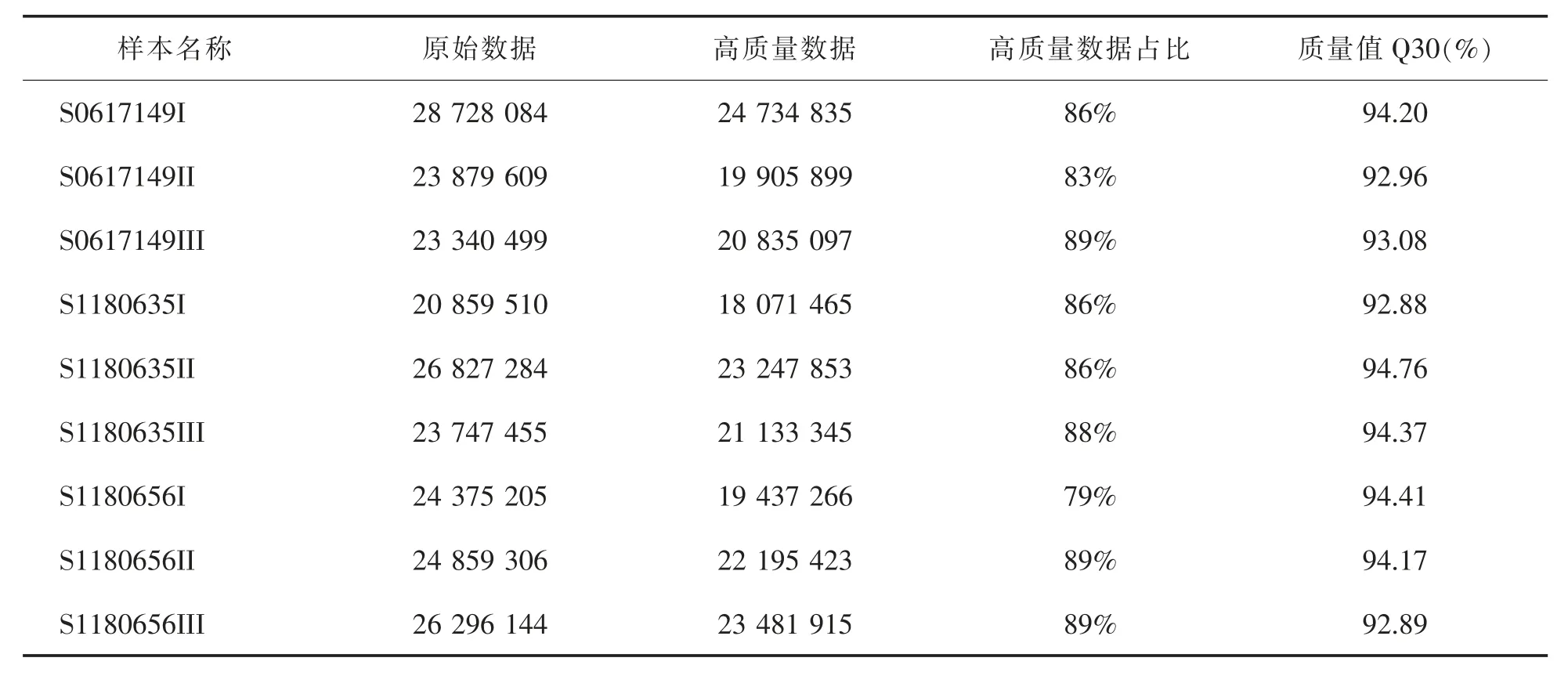
表3 转录组的数据统计
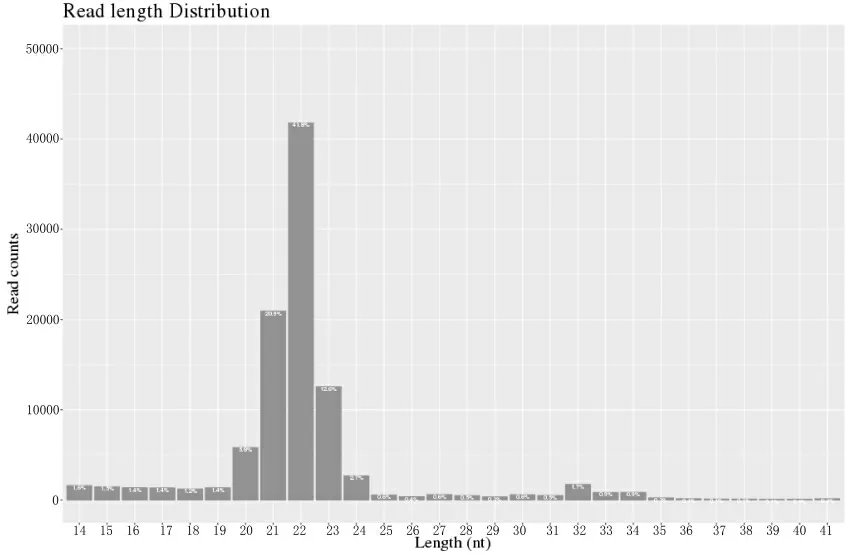
图1 S0617149I样本的reads长度占比情况
2.2.2 基因组比对结果
如表4,平均每个样本有8 173 966条reads比对到miRBase mature数据库上,平均每个样本可比对reads占比为37.90%;平均每个样本有18 544 011条reads比对到Genome数据库上,平均每个样本可比对reads占比为86.35%,73.09%在参考序列上只有唯一比对位置,占比越高说明转录组的测序数据利用率越高,可用于后续试验;平均每个样本有17 160 325条reads比对到Rfam数据库,未被注释的reads有2 116 948条,各类小RNA的分类注释和数量统计结果如表5,可用于后续新miRNAs的预测。

表4 比对到各基因组数据库的情况
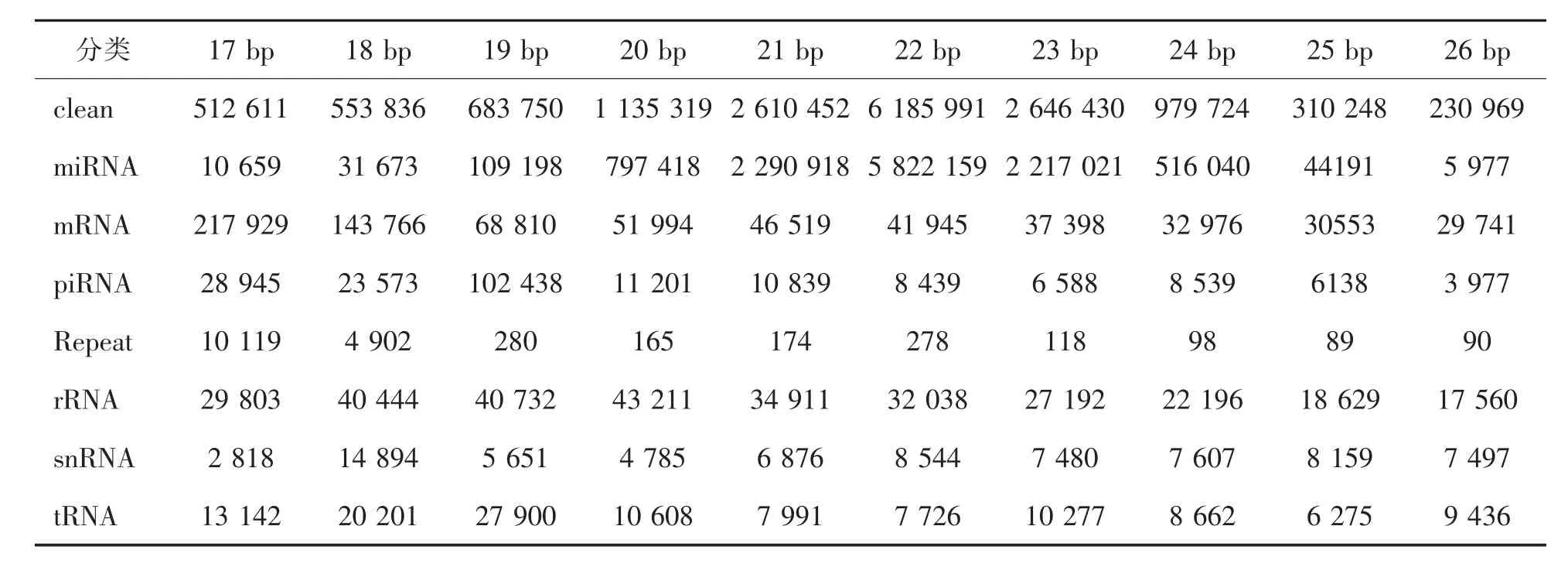
表5 不同长度的数据库读取映射
2.2.3 miRNAs鉴定结果
共鉴定了143个已知的miRNAs,并发现这些miRNAs属于75个家族,其中oar-let-7家族的最多,有7个:oar-let-7a、oar-let-7b、oar-let-7c、oar-let-7d、oar-let-7f、oar-let-7g、oar-let-7i,oar-miR-200家族的有3个:oar-miR-200a、oar-miR-200b、oar-miR-200c;并预测了759新miRNAs。
2.2.4 差异表达miRNAs的筛选
在已知的miRNAs中,相较于P1组,P1与P2两个时期共鉴定了62个差异表达miRNAs,全部下调;而在P1与P3两个时期鉴定了差异表达miRNAs 71个,包含P3时期3个显著上调的miRNAs与68个显著下调的miRNAs;在P2与P3时期未发现差异表达miRNAs,其中oar-miR-370-3p、oar-miR-541-3p、oar-miR-134-3p、miR-125b等miRNA在三个时期差异较明显;在新预测的759个miRNAs中,相较于P1组,P1与P2两个时期共鉴定了111个显著差异表达miRNAs,包含P2期7个显著上调的miRNAs与104个显著下调的miRNAs;而在P1与P3两个时期比较中鉴定了172个差异表达miRNAs,包含P3期25个显著上调的miRNAs与147个显著下调的miRNAs。相较于P2组,P3组共鉴定了71个显著差异表达miRNAs,其中包含23个显著上调的miRNAs与48个显著下调的miRNAs。以上结果表明休止期和生长期比较组获得的差异miRNAs明显多于其他两个比较组,说明由休止期向生长期转变过程中,有更多的miRNAs参与调控毛囊周期性变化过程;同时在三个比较组中,差异表达miRNA都是下调表达的比上调表达的多,说明在和田羊毛囊周期性变化过程中miRNA主要发挥负性调控作用。将上述数据绘制成火山图,如图2,为新预测的miRNAs中P2/P1对比组差异表达miRNAs的火山图。

图2 新预测miRNAs中P2/P1比较组差异表达miRNAs的火山图
为了进一步鉴定差异表达miRNA在和田羊3个不同时期中的分布情况,对已知的差异表达miRNAs进行了维恩图分析(图3),在维恩图分析中可直观展现出各差异比较组合之间共有与特有的差异表达miRNAs数量,在P2/P1时期检测到了6个时期特异性miRNAs:oar-miR-329a-5p、oar-miR-411b-3p、oar-miR-433-5p、oar-miR-496-3p、oar-miR-539-5p、oar-miR-665-5p,在P3/P2时期检测到了2个时期特异性miRNAs:oar-miR-134-3p、oar-miR-323a-5p。提示这些时期特异性表达miRNAs在毛囊周期性变化过程有重要作用。
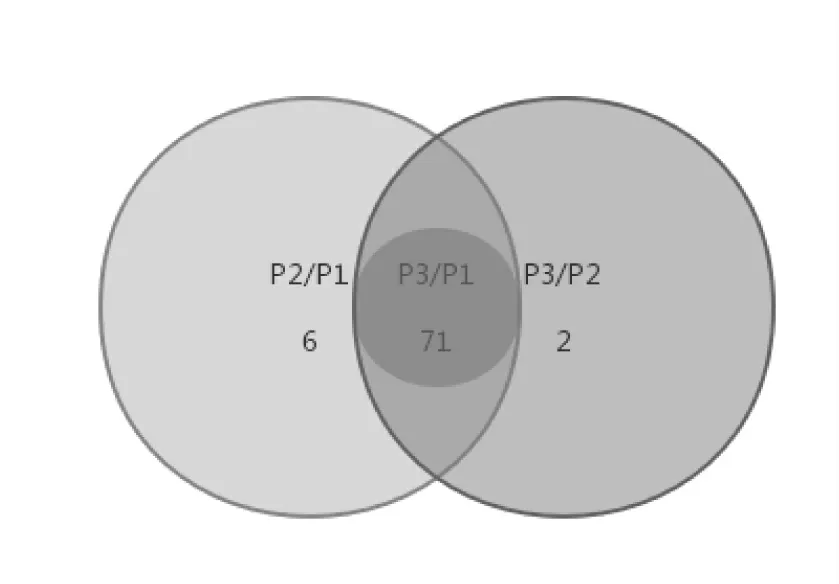
图3 已知的miRNA中差异表达miRNAs维恩图分析
2.3 靶基因预测
对上述筛选的和田羊毛囊周期性变化的3个时期的所有差异表达miRNAs分别使用miRanda、miRDB、TargetScan工具进行靶基因预测,取三者交集为差异表达miRNAs调控的靶基因。在生长期与退行期比较组173个差异表达miRNAs预测得到1 883个靶基因;休止期与生长期比较组243个差异表达miRNAs预测得到1881个靶基因;休止期与退行期比较组71个差异表达miRNAs预测得到140个靶基因。我们发现在这些预测得到靶基因中存在众多与毛囊生长、发育密切相关的功能基因,例如差异表达miRNA oar-miR-134-5p和oar-miR-432共同调控的KRT15已知在毛囊发育期、生长期启动和静止期的毛囊隆突区细胞表达,但在生长期和退化期表达不一致[16];差异表达miRNA oar-miR-495-5p调控的BMP4属于骨形态发生蛋白质(BMP),而BMP在控制皮肤生长、出生后的组织重塑和肿瘤发生上起着重要的作用,是毛囊发育所涉及的重要信号分子之一[17];oar-miR-433-3p和oar-miR-432的靶基因KRT25被发现可能与滩羊皮肤不同生长阶段的毛囊周期性变化有关[18]。以上预测的靶基因均与毛囊发育密切相关,提示调控这些基因的miRNAs可能通过靶向调控靶基因参与毛囊周期性变化过程,而其中确切的靶向关系也需要进一步的试验来进行验证。同时,我们还发现miRNAs与靶基因的调控关系可能是一一对应的,也可能是多个miRNAs共同调控单个靶基因,还可能是单个miRNA同时调节多个靶基因,如图4,oar-miR-485-3p既能调控STEAP4、INA、SMC4、FSD2,同时还和oar-miR-154a-3p共同调控GHITM。这说明和田羊中miRNAs具有多重靶向性。

图4 差异表达miRNAs与靶基因网络
2.4 靶基因GO和KEGG富集分析
本研究通过GO富集分析三个比较组的靶基因被注释毛囊发育相关的激素水平调节、细胞分解代谢、周期蛋白结合、骨细胞发育等GO条目中;通过和KEGG富集分析发现靶基因显著富集在与毛囊发育相关的AMPK、Hedgehog、TNF信号通路中,图5为退行期和生长期比较组靶基因KEGG富集结果中选取的显著富集的20个KEGG通路绘制成的柱形图。大量研究证实,出生后毛囊周期性变化激素调控,如雄激素以及雌激素等,并通过各相关调控信号分子/通路包括Wnt/β-catenin、骨形成蛋白(BMP)、Sonichedgehog(Shh)、notch、Hox、成纤维细胞生长因子(FGF)、胰岛素样生长因子(IGF)、血小板衍生生长因子(PDGF)、转化生长因子(TGF)、AMPK参与毛囊周期性发育过程。本研究与前人研究相符,提示在和田羊中皮肤毛囊差异表达miRNAs通过各信号通路调控靶基因参与毛囊周期进程。

图5 基于KEGG数据库的靶基因相关通路功能分布图
2.5 荧光定量PCR
结果如图6所示,所有选择的7个miRNAs表达趋势与转录组测序结果一致,说明转录组测序结果准确可靠。

图6 差异表达miRNAs qRT-PCR与转录组测序比较
3 讨论
和田羊尤其是山区型和田羊,其羊毛是其经济价值的所在,羊毛的品质决定着经济价值,羊毛的品质受毛囊的影响,而毛囊的发育和周期性变化受到多种因子的调控。很久之前就有研究证明miRNAs能够通过与mRNA的3'UTR序列配对的形式与mRNA结合,来影响mRNA的翻译过程,起到转录后的调控作用[19]。近年来有研究表明,miRNAs在多种哺乳动物皮肤毛囊中均有表达,在毛囊发育中起着重要作用[20]。为了探讨miRNAs在和田羊毛囊周期性变化中皮肤组织的转录组表达差异及可能发挥的作用,本研究利用高通量测序分析发现miRNAs在三个时期比较组有显著差异表达,表示miRNAs可能参与调控和田羊毛囊的周期性变化进程。
Liu[21]利用深度测序技术鉴定了绒山羊皮肤中的miRNAs,发现了22个全新的山羊miRNAs。张桂山[22]在辽宁绒山羊和乾华肉用美利奴羊皮肤毛囊发现毛囊发育的三个时期—生长期、退行期和休止期特有的miRNAs数分别为21个、23个和26个,15个、30个和27个。Yuan等[23]鉴定了绒山羊皮肤毛囊的399个保守的miRNAs,其中有36个miRNAs在毛囊发育的3个时期都有表达,有23个在皮肤毛囊发育的生长期特异表达,29个皮肤毛囊发育的退行期特异表达,44个在皮肤毛囊发育的休止期特异性表达。本研究共鉴定了143个已知miRNAs并预测了759个新miRNAs,在退行期和生长期对比组鉴定差异表达miRNA 173个,休止期和生长期对比组发现差异表达miRNA 243个;休止期和退行期对比组发现差异表达miRNA 71个;并发在退行期和生长期检测到了6个时期特异性miRNAs:oar-miR-329a-5p、oar-miR-411b-3p、oar-miR-433-5p、oar-miR-496-3p、oar-miR-539-5p、oar-miR-665-5p,在休止期和退行期检测到了2个时期特异性miRNAs:oar-miR-134-3p、oar-miR-323a-5p;而在三个时期显著差异表达miRNAs中的oar-let-7家族与山羊皮肤毛囊周期性生长发育密切相关[24],oar-miR-200家族在羊驼皮肤和毛囊发育中也可能起重要作用[25],并且发现显著差异表达miR-125b,可以导致皮肤增厚,对毛囊周期生长具有调节作用,但是有研究报道的miR-203对调控皮肤生长发育有重要调控作用,而在我们的研究中,在和田羊毛囊的3个时期对比组中均未发现miR-203的差异表达,说明出生后毛囊周期中的miRNA调控与胚胎期及出生后皮肤中的miRNA调控是存在差异的,还需要进一步研究。以上差异表达miRNAs可作为和田羊毛囊周期性变化过程中的关键候选miRNAs用于更进一步的研究。
为了探究差异表达miRNAs可能和田羊周期性变化中进程发挥的作用,筛选出各个时期差异表达的miRNAs之后,对差异表达miRNAs进行靶基因预测并进行功能富集分析。闫海龙等人[26]通过对毛囊干细胞与毛乳头细胞进行转录组和miRNAs测序分析,以及毛乳头细胞和毛乳头细胞来源外泌体进行的miRNAs测序分析鉴定了4个与毛囊干细胞增殖分化相关的关键候选miRNAs,分别为miR-1、miR-151-3p、miR-182、miR-143-3p,并发现let-7b-5p能够靶向调节预测的FOXC和MSI2的靶位点;江倩等人[27]发现miR-Let 7a在退行期的表达量高于生长期的表达量,并进一步证实miR-Let 7a可通过抑制两个靶基因Cmyc和FGF5的作用进而调控辽宁绒山羊的毛囊发育。Liu等[21]发现部分miRNA的靶基因可能通过Wnt信号通路参与调控绒山羊皮肤毛囊发育。本研究利用miRanda等软件对差异表达miRNAs的靶基因预测[28,29],发现KRT15是oar-miR-134-5p和oar-miR-432共同靶基因,oar-miR-495-5p靶基因是BMP4、oar-miR-433-3p和oar-miR-432有共同的靶基因KRT25。并对所有靶基因进行GO和KEGG富集分析,发现这些靶基因主要富集在毛囊发育相关的激素水平调节、细胞分解代谢、周期蛋白结合、骨细胞发育等GO条目中以及与毛囊发育相关的AMPK、Hedgehog、TNF信号通路中,说明miRNAs在毛囊周期性变化中可能通过这些通路来调节下游靶基因的表达来发挥调控作用,但miRNAs对预测的靶基因的具体靶向关系还需要进行更进一步的探究,需要进行更深层次的验证。
4 结论
本研究通过转录组测序获得了和田羊毛囊周期性变化中生长期、退行期、休止期的转录组文库,并对不同时期的转录组进行比较,鉴定了143个已知的miRNAs并预测了759个新miRNAs,进一步丰富了绵羊miRNA信息库;并在退行期和生长期比较组鉴定了173个差异表达miRNA,休止期和生长期比较组鉴定了243个差异表达miRNA,休止期和生长期比较组鉴定了71个差异表达miRNA,还通过靶基因预测和功能富集分析表明上述差异表达miRNAs可能参与和田羊毛囊周期性变化过程,但差异表达miRNAs及靶基因的互作调控关系还有待进一步验证。
猜你喜欢
科普童话·学霸日记(2021年9期)2021-09-23
成都体育学院学报(2021年1期)2021-07-16
西藏研究(2021年1期)2021-06-09
科教新报(2021年3期)2021-03-15
中等数学(2019年5期)2019-08-30
中等数学(2018年12期)2018-02-16
中国卫生(2016年12期)2016-11-23
健康必读(2016年8期)2016-09-26
系统工程与电子技术(2016年2期)2016-04-16
语言与翻译(2015年2期)2015-07-18
