
煤焦油中氯含量测定方法优化与实践
2021-12-10 05:35:28刘莹吕明张大奎王晓楠张欣
鞍钢技术 2021年6期
刘莹,吕明,张大奎,王晓楠,张欣
(鞍钢化学科技有限公司,辽宁 鞍山 114021)
煤焦油中氯含量的高低对焦油深加工工艺有重要影响。由于氯离子具有很强的穿透力,因而在煤焦油的运行环境下,氯离子将吸附于加氢压缩机等设备表面,迅速降低该处的pH值,酸化腐蚀设备和管道。煤焦油中的氯主要包括无机氯化物和有机氯化物。无机氯化物主要以氯化钠、氯化镁和氯化钙等碱金属或碱土金属盐等形式存在,可以通过脱盐脱水处理工艺去除;有机氯化物大多以氯代烷烃的形式存在。
国内外一般采用X-荧光测氯法、离子色谱法、微库仑法测定石油焦油和煤焦油中的总氯含量。X-荧光测氯法精密度高,但进样量大,成本高,而且X射线对操作人员的危害较大,较少用于煤焦油生产部门的检测作业。离子色谱法一般适用于水中氯含量的测定。微库仑法具有精密度高、样品适用范围广、操作简单等特点,适宜于煤焦油企业的生产批量检测。目前,鞍钢化学科技有限公司等国内企业均采用微库仑法测定煤焦油中的全氯含量,但在使用过程中经常出现一些问题,如滴定池平衡时间过长、基线漂移超出范围、检测器出现漂移记忆、裂解管积碳、多点绘制工作曲线线性不相关等,给检测工作带来了很多阻碍。本文对煤焦油中氯含量测定方法存在问题的原因进行了深入分析,并针对性地提出优化措施,通过实践解决了相关问题,实现了测定方法的优化。
1 测量仪器及测定方法
1.1 测量仪器与试剂
仪器采用EA5000测氯仪,包括进样器、裂解炉、石英裂解管、进样舟、屏蔽箱、气路传输系统、微量注射器(100 μL)等部分。
试剂采用氯标准物质(10 mg/L),超纯水,冰醋酸(GR),醋酸钠(CHCOONa)。
1.2 测定方法
1.2.1 测定原理
煤焦油样品由进样器带入裂解管,在高温下瞬间裂解汽化,形成的HCl气体与载气(氩气)混合后通过气体传输管进入电解池中,HCl与电解液中预电解得到的Ag反应,形成难溶的AgCl沉淀。反应后,电解液中的Ag逐渐减少,引起电极电位的变化,该变化量可由检测电极对(又称指示电极对)[Hg/HgSO/Ag]检测出来,经控制器接收处理并操纵电解单元,通过电解电极对变化的电极电位进行电位补偿,使电解液中的Ag浓度恢复到起始工作零点。通过记录电解产生Ag所需的电量,根据法拉第电解定律,即可求出试样的氯含量。
1.2.2 绘制标准曲线
采用单点制备曲线法,通过选择接近被测样品含量的不同浓度的氯标准物质制备标准曲线,仅对当批试样有效。
1.2.3 测量方法
(1)调整滴定池
保持滴定池及电极的清洁,不可有污染;每次使用前用电解液冲洗复合电极、银电极、铂电极,调整池中液面高度和电极位置;滴定池置于屏蔽箱中,调节搅拌速度,以电解液搅起旋涡为宜。
(2)调整测氯仪至操作状态
裂解管温度为1 050℃;气体流速,氩气100 mL/min,氧气 300 mL/min;电解池滴定平衡点3 000 mV;滴定漂移范围10~100 mV。
(3)测定氯含量
待炉温和气流稳定后,打开检测器,选定氯含量测定方法模块,点击手动滴定,使滴定池达到终点平衡,待记录器基线漂移稳定后即可进样分析样品中的氯含量。用100 μL微量注射器通过硅橡胶隔板将样品注入石英舟中。每次分析前,均需使用与待测样品氯含量相近的标准样品进行校正,调节滴定终点和基线漂移,得到煤焦油氯含量积分曲线,与标准曲线对比后得到氯含量测定数据;然后按照前述步骤进行空白试验,得到空白实验积分曲线,与标准曲线对比后得到氯含量测定背景数据;两次测定结果的差值,即为煤焦油样品中的氯含量测定结果。
2 存在问题及优化措施
2.1 滴定池平衡时间过长
滴定池平衡时间过长的原因一般包括复合电极隔膜堵塞、电解液不足、电解液污染、工作电极对中的银电极表面存在玷污或氧化层等。根据以上可能存在的问题,提出以下优化措施:
(1)开始检测前检查复合电极隔膜是否堵塞。电极隔膜堵塞一般由复合电极的电极液中的硫酸钠结晶导致,此时可使用25~35℃的蒸馏水对复合电极自内而外反复冲洗,直至复合电极恢复洁净状态。
(2)检查电解池内的电解液液位,使其不低于液位最低刻度线。
(3)检查指示电极对的平衡计数值,使其≤3 000 mV。指示电极对平衡指示计数值范围一般为1 000~10 000 mV。当进行终点滴定时,信号低于1 000 mV,需要手动加入少许电解液;信号高于10 000 mV甚至达到了60 000 mV,说明电解液有污染,则电解液需重新配置。电解液由醋酸钠配制,而醋酸钠一般以CHCOONa·HO的形式存在,由于电解液中不允许水的存在,故必须将醋酸钠中的结晶水去除,之后可使指示电极对平衡指示计数值≤3 000 mV。
(4)检查指示电极对中的银电极,观察其表面是否存在玷污或氧化层,可导致测量仪器信号计数值偏高,稳定时间过长等问题。银电极若存在玷污,可用洁净的滤纸将其擦拭干净;若存在氧化层,可用砂纸打磨,或用干净的刀片将包裹银棒的四氟乙烯少量切割,直至露出白色的银棒横切面。但银棒不可露出过多,否则易导致银棒被空气氧化,形成致密的氧化银表层,进而影响银棒的电解效率,降低电解液中Ag浓度,影响分析的精确度。
2.2 基线漂移超出范围
基线漂移超出范围的原因一般包括:气路系统漏气、试剂和气体不纯、操作环境差、滴定池温度过高或过低等。
2.2.1 气路系统漏气
针对可能存在的系统漏气问题,使用测氯仪软件自带的流量测试组件,对气路系统进行手动检查。图1为测氯仪的气路系统检测示意图。
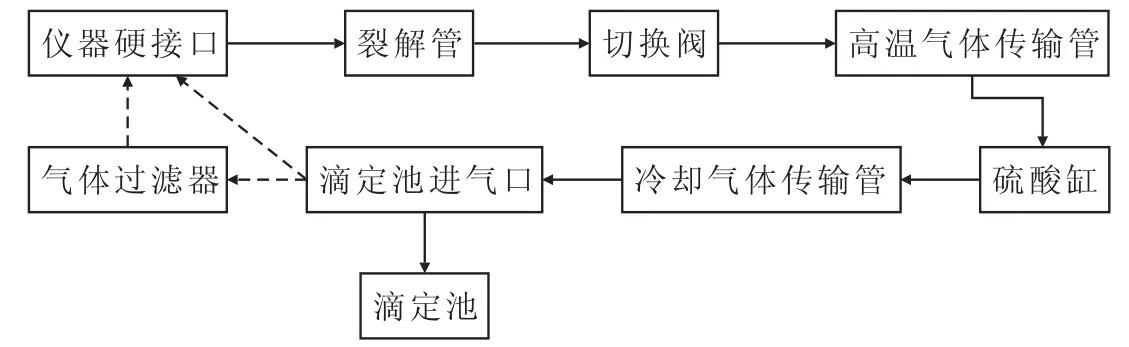
图1 气路系统检测示意图Fig.1 Schematic Diagram of Gas Path System Inspection
正常操作时,工作气体(包括氧气、氩气,不载入试样)由仪器硬接口接入,经裂解管、切换阀、高温气体传输管、硫酸缸、冷却气体传输管、滴定池进气口传送至滴定池进行滴定。
对气路系统进行检查时,首先清空硫酸缸中的硫酸,再将滴定池进气口通过软管和仪器硬接口短接,可测得气体流量值。然后在滴定池进气口与仪器硬接口之间加装气体过滤器,再测得气体流量值,前后两次测得的流量值之差≤5 mL/min,表明气体系统密封良好;否则,则说明气路系统存在漏气现象。解决措施包括检查各连接组件是否存在机械故障、更换易损零件等。
2.2.2 试剂和气体不纯
样品分析用浓硫酸必须为优级纯级(HSO含量98%),且必须每日更换。载气(氩气)和可燃气(氧气)纯度须达到99.99%以上。由于浓硫酸有很强的吸水性和氧化性,在移取硫酸过程中会吸收空气中的水分。因此,将浓硫酸注入硫酸缸后,需在敞口条件下通氩气曝缸20 min,可将浓硫酸中的水分和杂质去除,然后盖上玻璃球盖。
2.2.3 环境和滴定池的温度
实验室温度或滴定池池温过高或过低均可导致基线漂移不稳,滴定过程中电极亦不灵敏。图2为实验室温度过高时样品的氯含量谱图。
由图2可以看出,实验室温度过高时样品的氯含量谱图衍射峰特征明显不正常,无法开展正常测定。故配制后的电解液须在室温条件下放置,实验室操作最佳温度范围为23~25℃,测氯仪须避免阳光直射,不可置于通风处,保证供电正常,无干扰,避免仪器震动;滴定池的温度须低于实验室温度2℃。由于滴定池底座垫配有帕尔贴制冷片,只能降温不能升温,若滴定池温度过低,同样不能进行滴定,此时需将滴定池温度升至21~23℃方可继续操作。
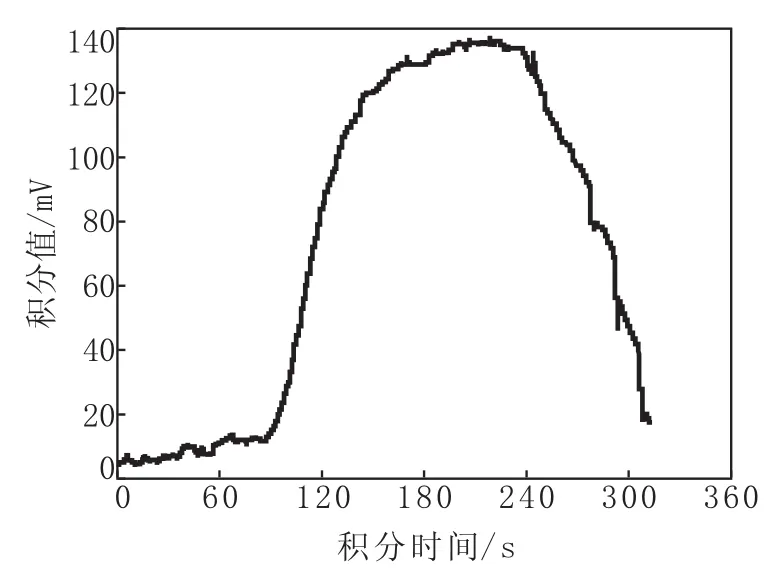
图2 实验室温度过高时样品的氯含量谱图Fig.2 Chlorine Content Spectrum of Samples when Temperature at Laboratory was TooHigh
2.3 检测器出现漂移记忆
仪器长期使用时经常存在漂移记忆现象。当测量平行样品时,两个平行样品测量的起始漂移必须尽量接近,漂移差值为0~90时,可保证平行样测定结果不会出现超差现象。因此,测量前观察并记录漂移数值,平行样品在进样时漂移要保持一致,以提高测量结果的精密度和准确度。
2.4 裂解管积碳
裂解管积碳是反应不完全的表征,会使转化率降低,并吸附被测样品,使被测样品测量结果偏小。进样量过大,氧气和氩气流量过小,二次燃烧吹扫时间不充分,进样垫破损后残渣掉入进样舟内随样品进入裂解管等均会导致裂解管积碳。解决措施:首先查看切换阀的过滤膜是否有污染,如果过滤膜呈黑色,说明裂解管积碳,需更换过滤膜。再检查裂解管,若有黑色或棕色的污染,可选用马弗炉或煤气喷灯高温除去;若裂解管内有白色或乳白色的附着物,则无法去除清洁,应更换裂解管。
图3是进样量过大或样品浓度过高时样品氯含量谱图。

图3 进样量过大或样品浓度过高时样品氯含量谱图Fig.3 Chlorine Content Spectrum of Samples when Charging Too Many Samples or Samples with Too High Concentration
由图3可以看出,该谱图出现多个异常平头峰。一般情况下,当被测样品中进样量过大或样品含氯量偏高时,有可能超过滴定池测定量程,此时反馈电信号超载,测得谱图易出现平头峰现象。此时可通过降低进样量、稀释样品或更换较低灵敏度的滴定池等措施予以解决。
2.5 多点绘制工作曲线线性不相关
由于测氯仪检测器灵敏度高,对作业环境要求也较苛刻,而现场作业条件有限,如噪音大、震动大等,导致多点绘制标准曲线很难达到线性要求,无法满足现场实际应用,故根据实际作业条件应采用单点校正法绘制标准曲线。主要包括通过选择接近被测样品含量的氯标准物质制备标准曲线,以及通过计算氯标准物质的绝对含氯量设计称样量。
单点校正法主要过程:先根据样品氯含量的范围选择对应的氯标准物质,然后对标准物质进行三次测定,当其中两次测定结果的变异系数≤3%时,则取该两次测定结果的平均值为基准绘制标准曲线。每批样品均需单独绘制标准曲线,以保证测定方法的精确度和准确度。单点校正法相对多点校正法还具有以下优势:适宜于测定浓度波动范围较大的样品,分析时间短,工作效率高,成本低,设备使用寿命长等。
图4是10 mg/L氯标准物质氯含量标准曲线和谱图。
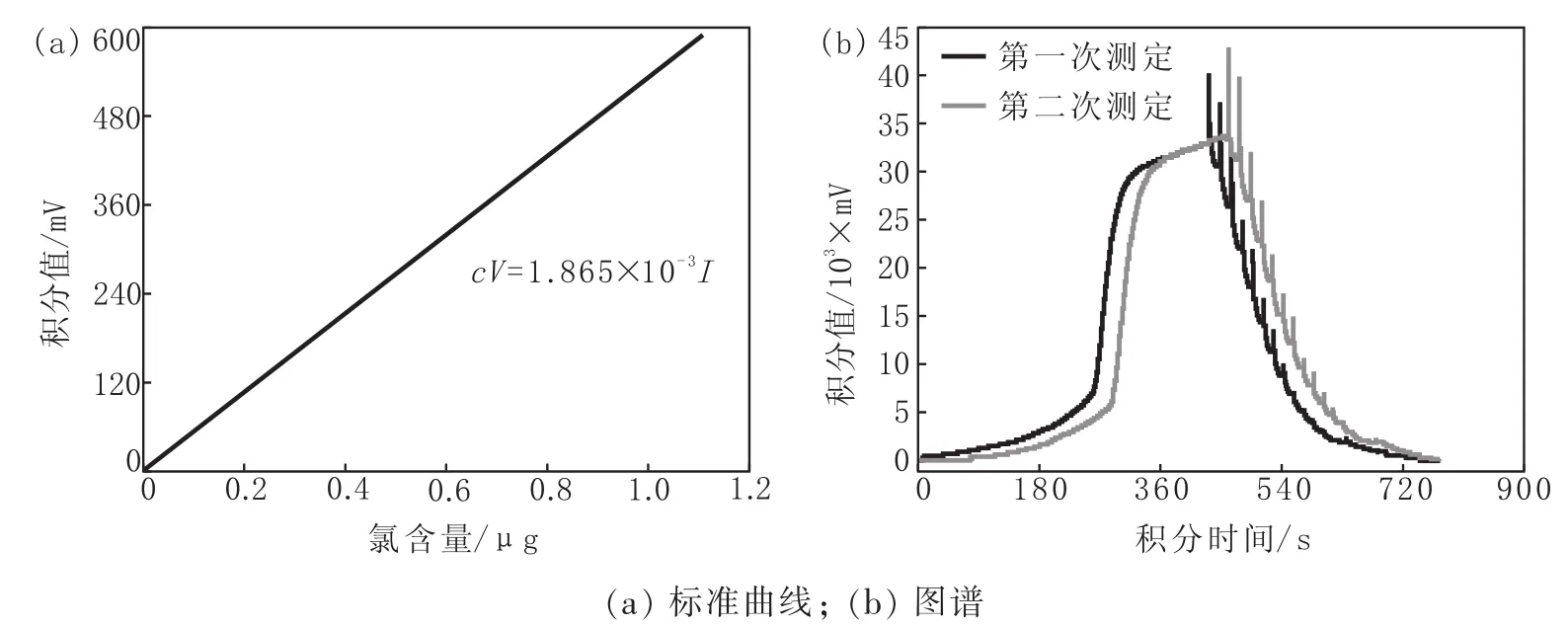
图4 10 mg/L氯标准物质氯含量标准曲线和谱图Fig.4 Standard Curve of Chlorine Content of 10 mg/L Chlorine Standard Substance and Its Chlorine Content Spectrum
按照单点校正法拟合出的10 mg/L氯标准物质氯含量标准曲线的公式为:

c
为氯标准物质的浓度,mg/L;V
为进样量,μL;I
为氯标准物质的积分面积,mV。由图4可以看出,两次标准物质测定结果的特征衍射峰明显突出,且无平头峰出现,谱图形貌整体完整,两次测定图谱形貌近似度较高,据此制定的标准曲线可满足精确度要求。
3 实践效果
鞍钢化学科技有限公司一回收作业区采用以上优化措施后,煤焦油全氯含量及其精密度分析结果如表1所示。由表1可以看出,优化后的相对标准偏差比优化前明显降低,由4.78%降至1.74%,满足测氯仪相对标准偏差≤3%的要求,说明优化后的测量方法的精密度和准确度均有明显提升。
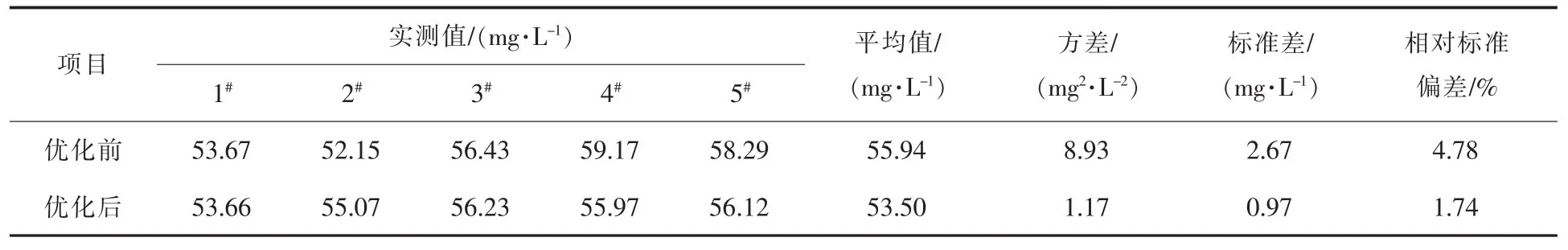
表1 煤焦油全氯含量及其精密度分析结果Table 1 Total Chlorine Content of Coal Tar and Its Precision Analysis Results
4 结语
鞍钢化学科技有限公司采用微库仑法、EA5000测氯仪测定煤焦油中氯含量,通过对滴定池平衡时间过长、基线漂移超出范围、检测器出现漂移记忆、裂解管积碳和多点绘制工作曲线线性不相关等问题进行分析,采取电极和电极隔膜清理、电解液和浓硫酸脱水处理、保证气体纯度和实验室温度、清除裂解管积碳、清除漂移记忆、采用单点校正法代替多点校正法等优化措施。实施后,有效提高了焦油氯含量测定的精密度和准确度,提高了工作效率,并延长了设备使用寿命,满足了作业现场使用要求,可为煤焦油氯含量分析相关人员提供借鉴。
猜你喜欢
云南化工(2021年6期)2021-12-21 07:31:00
航天控制(2020年5期)2020-03-29 02:10:34
山东冶金(2019年5期)2019-11-16 09:09:12
山东冶金(2018年6期)2019-01-28 08:14:50
机电元件(2018年4期)2018-08-09 12:17:38
电源技术(2016年2期)2016-02-27 09:04:59
中国资源综合利用(2016年7期)2016-02-03 03:00:19
橡胶工业(2015年2期)2015-07-29 08:29:44
化工管理(2015年12期)2015-03-24 21:06:07
振动、测试与诊断(2014年5期)2014-03-01 01:14:26
