
高效液相色谱-串联质谱法测定水中4种大环内酯类药物
2021-12-09 02:09:48杨创涛黄慧星
供水技术 2021年5期
杨 颖, 杨创涛, 黄慧星
(广东粤港供水有限公司, 广东 深圳 518000)
阿维菌素类药物(avermectins, AVMs)主要来源于链霉菌发酵产物的分离提取,属于大环内酯双糖类化合物,具有高选择性和高毒杀虫性,对各种农业害虫和动物寄生虫等有较好的驱杀效果,目前作为一种新型的农畜两用药应用较为广泛[1-2]。然而,该类药物极容易通过食物链富集,会导致运动和中枢神经系统毒性,当前世界卫生组织(WHO)以及包括中国在内的许多国家和地方组织等已将阿维菌素类药物确定为高毒性化合物,并对其残留量进行了限制[3-4]。水环境是农牧产业重要的排放受体之一,随着该类药物用量的不断增大,人类饮水安全的风险隐患也在逐步加剧,因此,加强对水环境中阿维菌素类药物的监测是十分有必要的。
阿维菌素类药物具有高分子量,较难气化,目前主要检测方法是液相色谱-光谱法(荧光法、紫外法)和液相色谱-串联质谱法,其中紫外法灵敏度低,干扰影响因素多,荧光法涉及衍生化处理,操作复杂,而高效液相色谱-串联质谱法具有高选择性和高灵敏度,目前应用日益广泛[5-6]。当前,我国已出台高效液相色谱-串联质谱法应用于饲料、粮食、蔬菜、肉禽等食品中阿维菌素类药物检测的标准方法[7-8],但水质相关的检测方法则较为少见。笔者采用超高效液相色谱-串联质谱法,同时测定水中阿维菌素、伊维菌素、多拉菌素、乙酰氨基阿维菌素,对基质相对简单的水质样品,仅需简单的固相萃取处理即可上机检测,测定下限可达0.60~0.80 μg/L,灵敏度高,抗干扰能力强,可实现这4种大环内酯类药物的快速、准确、高灵敏度检测。
1 实验部分
1.1 仪器与试剂
超高效液相色谱串联质谱仪(Waters OA Acquity UPLC TQD MS/MS);硅胶颗粒色谱柱(Waters ACQUITY UPLC HSS T3:50×2.1 mm,1.8 μm);氟苯基色谱柱(Waters ACQUITY UPLC CSH:50×2.1 mm,1.7 μm);亚乙基桥杂化颗粒色谱柱(Waters ACQUITY UPLC BEH C18:50×2.1 mm,1.7 μm);针头式过滤器(PTFE,0.45 μm)。
阿维菌素、伊维菌素、多拉菌素、乙酰氨基阿维菌素有证标准溶液,100 mg/L,甲醇介质;乙腈、甲酸、醋酸铵,色谱纯。
1.2 样品前处理
对于相对清洁的水样,直接取上清液进行固相萃取;对于浑浊水样,预先经0.45 μm滤膜过滤后,再进行固相萃取。
固相萃取步骤如下:依次用3 mL乙腈、10 mL水活化固相萃取小柱(Shimadzu InertSep PLS-3 PLS-3);取20 mL清洁水样,添加1%(体积比)乙腈,缓慢过柱后抽干小柱;然后依次使用0.5 mL乙腈进行2次洗脱;合并2次洗脱液后用乙腈定容至1.0 mL,然后添加1.0 mL水配制成(1+1,体积比)乙腈水溶液,混匀后转移入棕色进样瓶中待测。
1.3 色谱条件
采用Waters ACQUITY UPLC BEH C18色谱柱(50×2.1 mm,1.8 μm),柱温为35℃;以(0.05 mmol/L乙酸铵+0.1 %甲酸,体积比)水溶液为流动A相,以(0.1 %甲酸,体积比)乙腈为流动B相;进样体积为10 μL;采用梯度洗脱方式,梯度程序见表1。
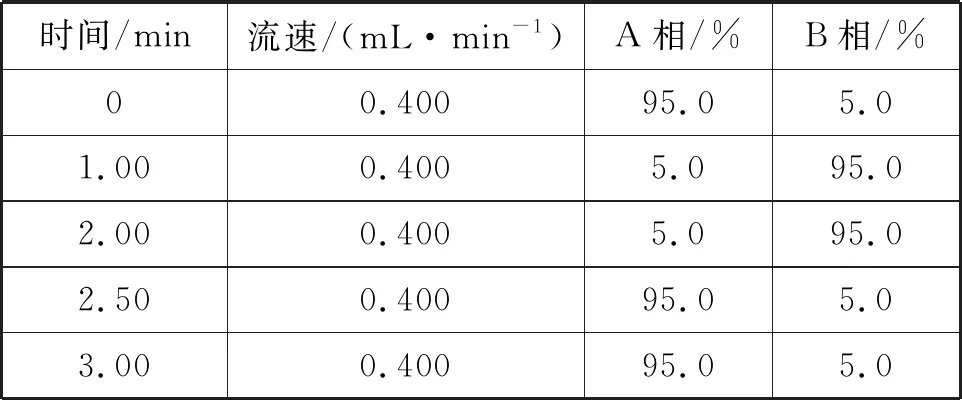
表1 液相色谱流动相梯度洗脱程序
1.4 质谱条件
离子源:ESI,负离子模式;毛细管电压:1.00 kV;离子源温度:120℃;脱溶剂气温度:400℃;脱溶剂气流量:600 L/h;反吹气流量:20 L/h;多离子反应监测方式(MRM),具体条件见表2。

表2 多离子反应监测条件
注:带*的为定量离子对,对于不同质谱仪器,参数可能存在差异,测定前应将质谱参数优化到最佳。
2 结果与讨论
2.1 固相萃取柱的选择
实验选择了Shimadzu GL WondaSep系列的C18、Shimadzu InertSep系列的PLS-3、Waters Oasis系列的HLB等3种不同材质的固相萃取小柱,配制浓度约为5 μg/L的样品进行富集萃取效果研究。
回收率结果表明,PLS-3小柱的效果明显优于C18和HLB小柱,使用PLS-3小柱萃取后4种目标药物的回收率都要高于其他2种小柱近2倍。因此,选定Shimadzu InertSep系列的PLS-3作为固相萃取小柱。
2.2 色谱柱的选择
实验研究了Waters ACQUITY UPLC HSS T3、Waters ACQUITY UPLC CSH Fluoro-Phenyl和Waters ACQUITY UPLC BEH C18 这3种不同色谱分析柱对4种目标化合物的分离效果。
结果表明,CSH色谱柱无法将4种目标物分离,且灵敏度偏低;BEH C18和HSS T3可以较好地将4种目标物分离,且BEH C18可获取更高的灵敏度。因此,选定Waters ACQUITY UPLC BEH C18作为色谱分析柱。
2.3 进样样品基质的选择
实验对比了纯水、(1+1,体积比)乙腈水和纯乙腈3种进样基质条件下的样品响应灵敏度。结果表明,以(1+1,体积比)乙腈水溶液作为进样样品基质有最高的灵敏度响应,纯水基质下几乎没有响应。因此,选定(1+1,体积比)乙腈水作为进样样品基质,该条件下样品经固相萃取后无需氮吹,可直接用乙腈和水定容,也提升了检测效率。
2.4 质谱条件的优化
实验中,以(0.05 mmol/L乙酸铵+0.1 %甲酸,体积比)水溶液和(0.1 %甲酸,体积比)乙腈为流动相,采用电喷雾离子源(ESI+),对4种目标化合物的质谱条件进行了优化。在确定各化合物的母离子后,采用子离子扫描方式对子离子进行优化选择,确定了定量离子和辅助定性离子对。通过优化毛细管电压、锥孔电压、透镜电压、碰撞能量及质谱分辨率等质谱参数,使3种目标化合物信号强度达到最佳。然后,进一步对离子源温度、脱溶剂气温度及流量、锥孔反吹气流量进行优化,使每种化合物的离子化效率达到最佳,最终确定目标化合物的多离子反应监测(MRM)条件,见表2。
2.5 目标物总离子流图
在实验选定的液相色谱和质谱条件下,4种大环内酯类药物(浓度均为50.0 μg/L) 都实现了有效分离,响应值良好,见图1。
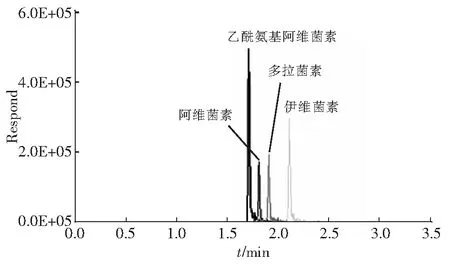
图1 4种大环内酯类药物的总离子流图
2.6 方法性能指标
2.6.1 线性关系、检出限和测定下限
采用经优化的分析条件,对不同浓度的标准溶液进行分析并绘制标准曲线。按照《环境监测分析方法标准制修订技术导则》(HJ 168—2010)对方法检出限测定的要求,采用低浓度空白加标水样(n=7)经固相萃取富集后上机测定,根据分析结果计算其标准偏差SD,检出限MDL=3.143×SD,定量限为检出限的4 倍。从表3可以看出,方法的线性相关性好,目标物检出限在0.10~0.20 μg/L,测定下限在0.40~0.80 μg/L,可满足水体低浓度样品的检测需求。
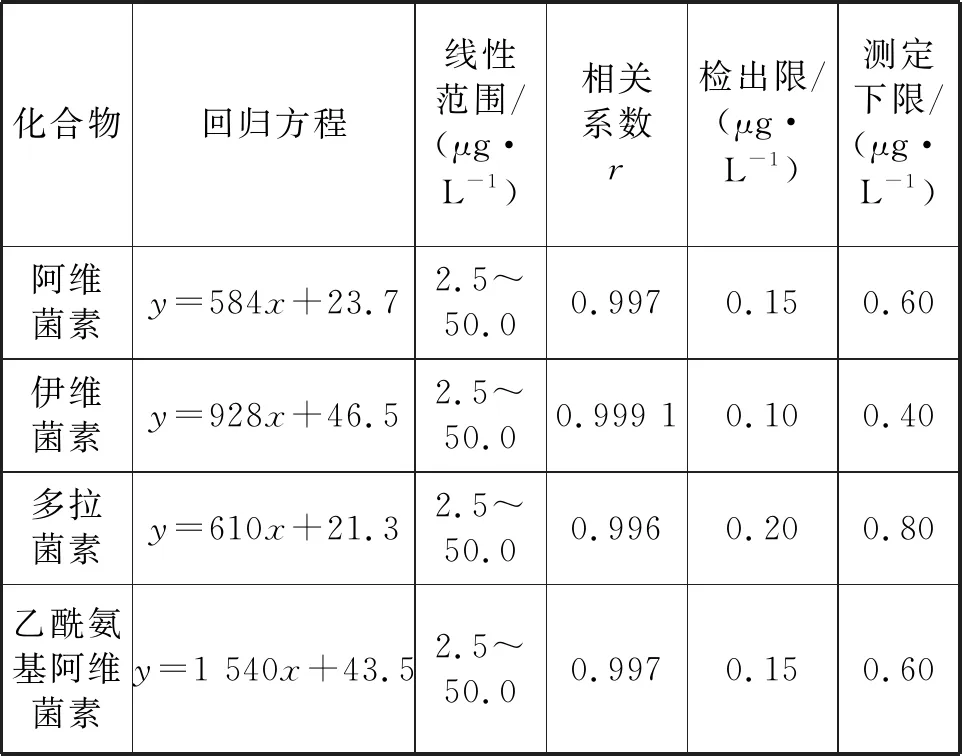
表3 4种大环内酯类药物的线性关系、检出限和定量限
2.6.2 精密度和准确度
对地表水、饮用水2种不同类型的实际水样中目标化合物的浓度进行测定,结果均为未检出。同时,在上述2种类型样品中分别加入不同浓度水平的标准物质,进行低、中浓度水平的加标回收实验,每个浓度分别平行配制7份样品,同时计算相对标准偏差和平均加标回收率。结果表明,平行样间相对偏差在5.6%~12.9%,水样的加标回收率在84.6%~126%,方法精密度和准确度良好。
3 结论
建立了超高效液相色谱质谱法测定水中阿维菌素、伊维菌素、多拉菌素、乙酰氨基阿维菌素等4种大环内酯类药物的分析方法。该方法的样品固相萃取前处理简单,有机溶剂用量少,环境友好,检出限、精密度和准确度令人满意,能够满足水体中大环内酯类药物的检测要求。
猜你喜欢
科学导报(2023年77期)2023-11-08 00:27:03
少年文艺(2022年8期)2022-07-08 10:02:47
农药科学与管理(2019年6期)2019-11-23 08:17:12
环境保护与循环经济(2017年4期)2018-01-22 03:27:11
临床医药文献杂志(电子版)(2017年12期)2017-05-18 01:28:00
中国经济周刊(2017年6期)2017-03-21 00:59:27
读写算·高年级(2016年3期)2016-05-30 01:53:46
兽医导刊(2016年4期)2016-04-05 12:35:02
浙江中西医结合杂志(2016年2期)2016-01-25 02:49:19
中国医疗美容(2015年1期)2015-07-12 10:06:18
