
内分泌干扰物对核受体二聚化影响的研究进展
2021-12-07 11:08:14黄付晏陈钦畅谭皓月郭婧于南洋史薇于红霞
生态毒理学报 2021年4期
黄付晏,陈钦畅,谭皓月,郭婧,于南洋,史薇,于红霞
污染控制与资源化研究国家重点实验室,南京大学环境学院,南京 210023
核受体(nuclear receptor, NR)超家族是由天然激素调控的转录因子(transcription factors, TFs),在细胞分化、发育、增殖以及代谢中发挥着重要作用[1]。环境中存在大量小分子有机化合物,如双酚A(bisphenol A, BPA)、羟基化多溴联苯醚(hydroxylated polybrominated diphenyl ethers, OH-PBDEs)、多氯联苯(polychlorinated biphenyls, PCBs)、多环芳烃(polycyclic aromatic hydrocarbons, PAHs)和杀虫剂等,它们会通过模仿或拮抗天然激素,靶向NR,进而产生内分泌干扰效应,导致不良健康影响[2-4],这种化合物被称为内分泌干扰物(endocrine disrupting chemicals, EDCs)。EDCs会导致人类产生严重的生殖发育疾病,如癌症[5]、心血管疾病[6]、肥胖[7]及生殖异常[8]等。据统计,欧盟EDCs相关疾病的治疗费用达到欧盟内部生产总值的1.28%[9],美国达到2.33%[10]。
有害结局路径(adverse outcome pathway, AOP)的概念被用来描述内分泌干扰效应。当EDCs作用于核受体后,会诱导分子启动事件(molecular initiating events, MIEs)的发生,即EDCs首先与核受体的配体结合口袋(ligand binding pocket, LBP)相结合,模拟或拮抗内源性激素配体与核受体结合形成核受体-化合物复合物,进而该复合物转移进入细胞核内形成同源二聚体(homodimer)或异源二聚体(heterodimer),结合到DNA反应原件(DNA-responsive element, DRE)上,通过招募共激活因子(coactivator, COA)或共抑制因子(corepressor, COR)启动或抑制转录[11-12]。MIEs反映EDCs与靶标核受体间的相互作用,其进一步引起细胞层次、器官层次等一系列关键事件(key events, KEs)的变化,最终导致内分泌干扰效应。目前对于EDCs分子启动机制的研究主要针对EDCs与核受体的结合过程,忽视了其对核受体二聚化过程的影响,而该过程的阻断可直接导致转录失活[13]。因此,本文从EDCs介导的核受体二聚化转录机制、核受体二聚化与转录活性间的关系以及基于活细胞的核受体二聚化研究方法3个方面对EDCs对核受体二聚化的影响进行了概述。
1 核受体转录机制(Transcription mechanism of nuclear receptor)
1.1 人类核受体家族
人类存在48个核受体(表1),基于其配体以及二聚化特征可将它们分为3类:(1)I类,也被称为类固醇核受体(steroid nuclear receptor),包括雌激素受体(estrogen receptor, ER)、雄激素受体(androgen receptor, AR)等。类固醇核受体主要以同源二聚体结构调控下游的转录过程[14-15]。其中,ER存在2种亚型,ERα和ERβ,可分别形成ERα同源二聚体、ERβ同源二聚体和ERα-ERβ异源二聚体调节转录[16]。(2)II类,也被称为非类固醇核受体(non-steroid nuclear receptors),包括甲状腺激素受体(thyroid hormone receptorα/β, TRα/β)、维甲酸受体(retinoic acid receptorα/β/γ, RARα/β/γ)以及组成型雄烷受体(constitutive androstane receptor, CAR)、类法尼醇X受体(farnesoid X receptorα/β, FXRα/β)等。该类核受体主要与维甲酸X受体(retinoid X receptorα/β/γ, RXRα/β/γ)形成异源二聚体调节下游基因的转录表达,如RAR-RXR[17]、FXR-RXR[18]和CAR-RXR[19]等。(3)Ⅲ类,也被称为孤儿核受体,其内源性配体还未发现或一部分核受体不存在配体,包括小异二聚体伴侣(short heterodimeric partner, SHP)、睾丸受体(testicular orphan receptor 2/4, TR2/4)等。这类核受体多数可以以单体或同源二聚体的形式结合各自的DRE来调节转录的表达。特别地,NR相关因子(nur-related factor 1, NURR1)可与RXR形成异源二聚体,并能被RXR配体结合激活,进而调节转录过程[20-24]。
1.2 核受体二聚体
已发表的大量结构化和功能化数据证实,核受体的DNA结合域(DNA binding domain, DBD)和配体结合域(ligand binding domain, LBD)与二聚过程高度相关[25]。由于DBD贡献的二聚化界面很小[26],而LBD贡献很大的二聚化界面,能够提供更大的二聚体稳定性,因此人们普遍认为LBD对二聚过程存在决定性的作用[27]。
1.2.1 典型核受体二聚体
核受体主要以同源二聚体或异源二聚体的形式调节转录。表1中3种类型的同源二聚体或与RXR形成的异源二聚体形式被认为是典型二聚体(typical dimers)[28]。部分典型异源二聚体存在的一个固有特征是既可被自身配体直接激活,又可被RXR的配体介导激活。因此,基于此特性可将典型异源二聚体进一步分为2类,第1类可被自身配体或者RXR配体激活,第2类只能被自身配体激活而RXR沉默[29-30]。特别地,对于第1类异源二聚体而言,如PPAR、CAR和LXR,当核受体自身的内源配体和伴侣RXR的配体同时存在时能产生协同效应[31]。
通过结晶实验,大部分典型核受体二聚体已有基于LBD的二聚体结晶结构(https://www.rcsb.org/)。通过对不同核受体二聚化结晶体结构进行研究,我们发现不同核受体产生的二聚体构象之间存在显著差异(图1)。
1.2.2 典型核受体二聚体的构象差异
多数典型核受体二聚体会由每个单体(monomer)的第9号α螺旋链(Helix 9)、第10号α螺旋链(Helix 10)和11号α螺旋链(Helix 11)互相接触构成二聚化界面(图1(a)、(b)和(c)),这种被称为经典二聚体结构[32-34]。有趣的是,一些核受体如AR、糖皮质激素受体(glucocorticoid receptor, GR)、盐皮质激素受体(mineralocorticoid receptor, MR)、孕激素受体(progesterone receptor, PR)等的二聚化方式与这种经典的二聚体完全相反(图1(d)和(e))[35-37],呈现出2个单体的5号α螺旋链(Helix 5)头对头的松散型二聚体构型。根据欧洲蛋白数据库(EMBL-EBI)的PDBePISA模块(https://www.ebi.ac.uk/pdbe/pisa/)提供的核受体界面信息发现,经典结构的二聚化界面能贡献更大的界面面积以及更多的相互作用。如ERα-ERα同源二聚体拥有高达14.561 nm2的二聚化界面面积,RAR-RXR和CAR-RXR异源二聚体界面面积分别达到11.963 nm2和12.112 nm2,而AR-AR、GR-GR同源二聚体界面面积分别只有10.003 nm2和8.095 nm2。氢键(hydrogen bonds)和疏水相互作用(hydrophobic interactions)是形成二聚界面的2种主要作用力。对氢键进行分析发现,经典二聚体构型,如ERα-ERα同源二聚体、RAR-RXR和CAR-RXR异源二聚体界面氢键数分别达到10、19和12个,而松散型二聚体AR-AR、GR-GR同源二聚体界面氢键只有5个,氢键数目越多表明界面相互作用越强。而界面溶剂化自由能(solvation free energy,ΔG)越负,表明核受体二聚体界面的疏水接触越多,通过对多种二聚体的ΔG分析发现,不同核受体产生的同种类型的二聚化构型之间也存在明显的差异性。例如,对于结构紧密的经典型二聚体,ER-ER同源二聚体界面ΔG为-59.9 kJ·mol-1,具有很强的疏水相互作用,而CAR-RXR异源二聚体界面ΔG只有-14.2 kJ·mol-1。而对于结构松散型二聚体,GR-GR同源二聚体界面ΔG达到-46.9 kJ·mol-1,AR-AR同源二聚体界面只有-12.1 kJ·mol-1(图1和表2)。
1.2.3 非典型异源二聚体
除了与RXR伴侣生成典型核受体异源二聚体以外,有大量研究表明,存在非典型异源二聚体(atypical heterodimer),即核受体并不与RXR伴侣异源结合形成二聚,如AR-GR[38]、GR-MR[39]、GR-PPAR[40]、ER-AR[41]、ER-GR[42]和PPAR-ERR[43]等。这些非典型异源二聚体不像典型异源二聚体结构被详细解析,并且可能局限于特定的细胞类型和体内生理条件,具有寿命短、瞬时性等特点,但其短暂的存在仍可能会对靶细胞或组织中基因表达产生强烈影响。因此,非典型核受体异源二聚体的生理相关性识别对配体调节核受体作用的现有知识提出了挑战[28]。

表1 人类核受体家族Table 1 Human nuclear receptor family
1.3 核受体二聚体转录机制
3类核受体经过二聚后会产生3类转录机制,具体如下:
Ⅰ类:类固醇核受体通常以单体形式或与伴侣蛋白(chaperones/cochaperones)形成复合体稳定存在于细胞质中,但在配体结合后,分子伴侣解离,核受体-配体复合物转移到细胞核中[44](以GR为例,图2(a))。值得注意的是,一些核受体(如AR、GR和MR)必须在配体结合后才能与分子伴侣解离以转移到细胞核中,而一些核受体(如ER、PR)可在无配体结合情况下转移到细胞核中[45-46]。进入细胞核后,这类核受体会形成同源二聚体,与其DRE结合,该反应元件为反向完全或不完全回文结构,后通过招募共激活因子或共抑制因子以促进或抑制转录[47]。

表2 部分核受体二聚化界面参数Table 2 Interface parameters of dimerization of partial nuclear receptors
Ⅱ类:非类固醇核受体,这种核受体通常和RXR形成异源二聚体,不管是否存在激活配体,均存在于细胞核中。无配体结合时与共抑制因子形成复合物,激活配体结合后,共抑制因子解离,与共激活因子结合[48]。这种与RXR形成的异源二聚体结合的DRE为直接重复结构[49](以RAR为例,图2(b))。
Ⅲ类:孤儿核受体,这种核受体无激活配体时存在于细胞核中,其作用机制与第Ⅱ类核受体一致,区别就是该类核受体可自身形成同源二聚体与直接重复结构的DRE结合[50](以VDR为例,图2(c))。
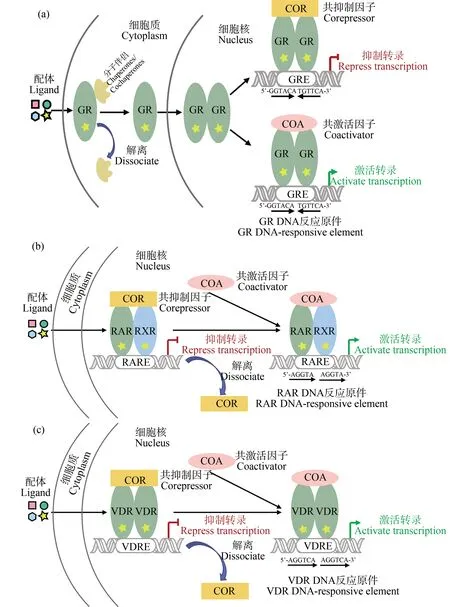
图2 核受体转录机制注:(a)GR-GR同源二聚体转录机制;(b)RAR-RXR异源二聚体转录机制;(c)VDR-VDR同源二聚体转录机制。Fig. 2 Transcription mechanism of nuclear receptorNote: (a) Transcription mechanism of GR-GR homodimer; (b) Transcription mechanism of RAR-RXR heterodimer; (c) Transcription mechanism of VDR-VDR homodimer.
2 内分泌干扰物对核受体二聚的影响(Effect of endocrine disruptors on dimerization of nuclear receptors)
已经有大量研究表明,EDCs与核受体的竞争结合过程和共因子招募过程对核受体最终的转录活动存在关键性作用[51-52]。但现有研究大都仅通过上述2个过程解释内分泌干扰活性存在假阴性或假阳性的结果[53](https://actor.epa.gov/edsp21/),而有研究表明在考虑核受体的二聚过程后能够降低这种误差[54]。因此,研究内分泌干扰物对于核受体二聚化作用的影响对于完善核受体介导的内分泌干扰事件的分子机制以及准确评估内分泌干扰物的效应具有重要意义。
2.1 内分泌干扰物可引起核受体不同形式的二聚化
通过检索ToxCast和Tox21数据库中体外实验的结果,发现针对ER做了比较完善的研究,包括化学物质对ER竞争结合(NVS_NR_hER)和ER二聚化的测定,其中对ER二聚化的测定包括ERα-ERα同源二聚体(OT_ER_ERaERa_0480、OT_ER_ERaERa_1440)、ERα-ERβ异源二聚体(OT_ER_ERaERb_0480、OT_ER_ERaERb_1440)以及ERβ-ERβ同源二聚体(OT_ER_ERbERb_0480、OT_ER_ERbERb_1440)。数据库中有多达227种EDCs物能够影响ER二聚化(表3)。
不管在激动剂配体还是拮抗剂配体存在的条件下,ER都可进行二聚化[27]。然而,配体所诱导ER二聚化的类型是不同的,相比于ERα-ERα同源二聚体(6.09%~7.38%的活性率),EDCs更易诱导ERα-ERβ异源二聚体(11.25%~12.22%的活性率)和ERβ-ERβ同源二聚体(10.02%~11.69%的活性率)(表3)。而有研究表明ERα和ERβ在调节雌激素作用中起相反作用,ERα-ERα同源二聚体促进激素依赖的乳腺癌细胞增殖,ERβ-ERβ同源二聚体对其起抑制作用,而ERα-ERβ异源二聚体在生物体中的作用尚不清楚[55-56]。Powell和Xu[56]发现植物雌激素染料木黄酮(genistein)、甘草黄素(liquiritigenin)可选择性诱导ERα同源二聚体、ERβ同源二聚体和ERα-ERβ异源二聚体的产生,Coriano等[55]研究了12种类黄酮化合物对于ER二聚体的差异性诱导。
一些研究也表明EDCs能够诱导其他核受体不同类型的二聚体形式。Depoix等[57]研究发现维生素D能够促进VDR与RXR的异源二聚。Putcha等[58]研究发现了甲状腺激素(3,5,3’-triiodothyronine, T3)和维甲酸(9-cis retinoic acid)在诱导TR-RXR异源二聚体间的负协同作用。Collingwood等[59]证明甲状腺激素能够促进TRβ和RXR的异源二聚。研究EDCs对不同类型二聚体的诱导对于研究内分泌干扰物转录活性的生理学相关性具有重要意义。
2.2 二聚体与转录激活的关系
分析OECD报告(Series on Testing and Assessment No. 309; http://www.oecd.org/officialdocuments)中具有体外(invitro)和体内(invivo)雌激素活性的14种参考化学品,其体外和体内活性数据来自美国环境保护局(US Environmental Protection Agency, US EPA)开展的EDCs筛选项目(Endocrine Disruptor Screening Program, EDSP)中第一阶段测试(Tier 1)的体外试验和体内子宫营养试验。将参考化学品的活性数据和Toxcast和Tox21数据库中数据对比研究发现,对于雌二醇、雌酮、己烯雌酚、5α-二氢睾酮、双酚A、双酚B、壬基酚、甲氧氯和滴滴涕等14种参考化学品,具有100%的二聚活性,但只有85.7%具有竞争结合活性(表4)。4-壬基酚、2,4’-滴滴涕这2种物质,其同时具有体外和体内雌激素活性,但不具有竞争结合活性,由此来看通过竞争结合活性预测化学物质活性存在假阴性,结合二聚活性能够更好地预测ER雌激素活性。Delfosse等[60]在研究双酚A(bisphenol A, BPA)、双酚C(bisphenol C, BPC)和双酚AF(bisphenol AF, BPAF)的雌激素效应的过程中发现,ERα同源二聚体晶体中两边单体内BPA的结合模式与天然雌激素雌二醇(estradiol, E2)类似,BPC的结合模式与抗雌激素他莫昔芬(tamoxifen, OHT)类似,有趣的是BPAF在2个单体中的结合模式相反,一个类似E2,另一个类似OHT,这种差异可能表明不同的配体对于核受体二聚化的变构调节,从而为药物设计提供了新的视角。同源二聚体的2个单体之间存在调节串扰,配体与一个单体的结合会调节二聚体中另一个单体的构象[61]。Judson等[62]使用包含ER配体结合、二聚化、转录激活和细胞增殖测定等在内的16个体外测试开发模型以预测1 811种化学物质的雌激素活性,发现基于至少4~7个体外实验的亚组的预测准确性和16个测试建立的模型相当,这些亚组模型的主要区别在于采用的二聚化测定结果不同。
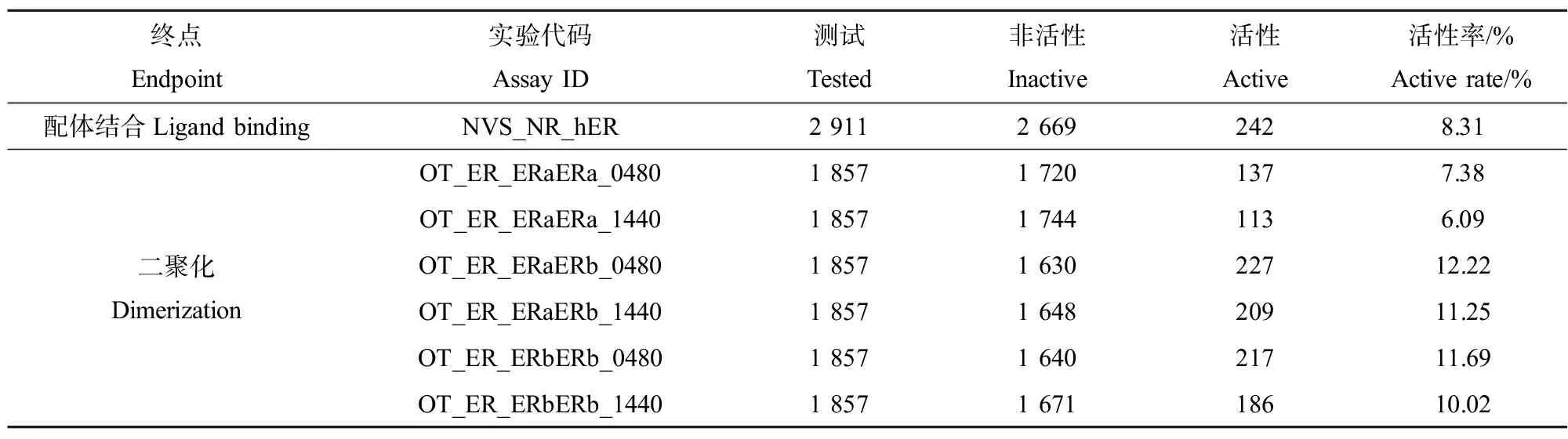
表3 针对雌激素受体的Toxcast和Tox21体外竞争结合和二聚化测试结果Table 3 In vitro ligand binding and dimerization test results of ToxCast and Tox21 for estrogen receptor
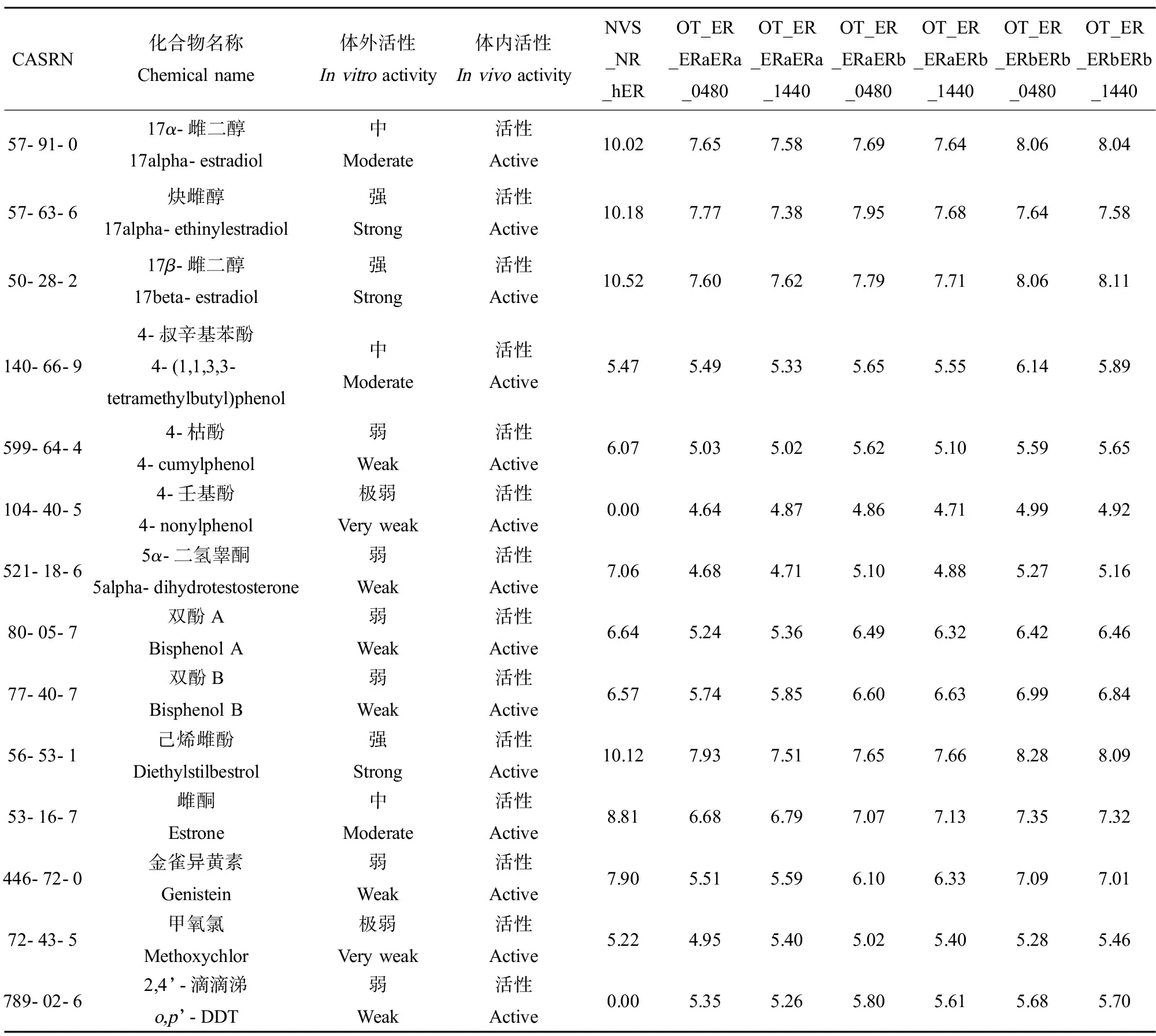
表4 参考化学品的体外、体内雌激素活性及其二聚活性Table 4 Dimerization activity of reference chemicals and its estrogen activity in vitro and in vivo
Nadal等[35]测试了配体对于诱导AR二聚化的影响,发现激动物质能够诱导AR二聚化,拮抗物质则不能。Depoix等[57]研究发现RAR激动剂能够显著增加RAR-RXR的异源二聚,而RAR拮抗剂能够抑制RAR-RXR的异源二聚。核受体二聚化与其转录活性的显著相关性表明在预测EDCs诱导核受体转录活性时必须考虑其对核受体二聚化的影响。
除了配受体结合、共因子招募这2个过程,考虑核受体二聚化过程对于研究内分泌干扰效应的产生至关重要。然而目前有关EDCs对核受体二聚化的影响,只对ER进行了相对比较完善的研究,包括EDCs对于2种亚型(ERα和ERβ)各自的同源二聚体以及其一起形成的异源二聚体的影响。但EDCs对于其他核受体同源或者异源二聚体形成的影响研究相对较少,且主要集中研究各自的内源性配体化合物或者相关药物,今后可加强研究EDCs对除ER以外的其他核受体的二聚化影响。
3 核受体二聚化的研究方法(Research methods of nuclear receptor dimerization)
检测同源或异源二聚体间蛋白-蛋白相互作用对于揭示核受体调控机制至关重要。目前,研究蛋白-蛋白相互作用的实验种类繁多,如基于蛋白结构解析的核磁共振(nuclear magnetic resonance spectroscopy, NMR)实验和X射线衍射(X-ray)实验,该类方法条件严格且价格昂贵[63];基于蛋白的免疫共沉淀技术(co-immunoprecipitation, co-IP)和蛋白质微阵列技术(protein microarrays),操作过程繁杂,无法检测到瞬时作用,且无法在活细胞中测定[64];另一种经典的测试技术是酵母二杂交或三杂交(Y2H/Y3H)试验,由于其简单易用,该体内技术被广泛用于大规模检测蛋白-蛋白相互作用,然而Y2H/Y3H常因酵母细胞中异种基因的表达而产生假阳性或假阴性结果[65]。为了克服以上缺陷,开发了一系列体内检测技术用于检测活细胞中的蛋白-蛋白相互作用,如荧光共振能量转移技术(fluorescence resonance energy transfer, FRET)、生物发光共振能量转移技术(bioluminescence resonance energy transfer, BRET)和生物分子荧光互补技术(bimolecular fluorescence complementation, BiFC)等,这些技术克服了非活体状态分析蛋白间相互作用的局限性[66],研究活细胞内核受体二聚化的形成对包括信号传导和转录调节在内的多种细胞过程具有重要意义[56]。然而,由于体外细胞筛选大量的干扰物费时费力,计算机辅助的核受体二聚化研究应运而生。
3.1 荧光共振能量转移(FRET)
FRET技术的供体分子(donor)和受体分子(receptor)均为荧光蛋白(fluorescent protein, FP,如YFP),其分别偶联2个目标蛋白,当供体和受体分子间距离<10 nm时,处于激发态的供体荧光团通过偶极子间的相互作用将能量以非辐射的方式转移给邻近的受体分子,即产生能量转移[67](图3(a))。
Feige等[68]利用FRET研究发现PPAR在没有配体的情况下很容易与RXR形成异源二聚体。Tamrazi等[27]以位点特异性方式用单个荧光团对ER进行化学标记,基于FRET测量ERα-LBD二聚体的热力学和动力学稳定性,并将该方法用于评估ER配体的激动剂和拮抗剂活性。Schaufele等[69]利用FRET研究了分别用CFP和YFP标记的AR二聚体的形成,发现AR的二聚化只发生在小分子配体结合后,并主要在细胞核内。Nadal等[35]利用FRET测试了配体对于诱导AR二聚化的影响,发现激动物质能够诱导AR二聚化,拮抗物质则不能。Yoshimura等[70]将磷酸化突变体RXRα(YFP-T82D/S260D-RXRα)与CFP-RARβ共转染于HEK293T细胞,在细胞核中检测到的FRET信号非常低,而未磷酸化的RXRα(YFP-T82A/S260A-RXRα)与CFP-RARβ转染后的FRET效率与野生型RXRα相当,表明磷酸化的RXRα丧失了与RXRβ异源二聚化的能力。
FRET具有高灵敏性和特异性,能够提供NR相互作用的位置信息,并且能够和显微镜、色谱技术等多种仪器和技术结合使用[71〗。但是FRET需要一对FP分别作为供体蛋白和受体蛋白,目前可用于FRET的供-受体对存在光漂白的缺陷,即供体的发射光谱和受体吸收光谱有明显的重叠[72-73]。
3.2 生物发光共振能量转移(BRET)
与FRET不同,BRET利用生物发光酶如海肾萤光素酶(renilla reniformis luciferase, Rluc)作为供体分子,荧光蛋白作为受体分子,当两者之间距离<10 nm时,供体分子在底物如腔肠素(coelenterazine)的催化下发光并将能量转移给受体[73](图3(b))。
Mulero等[74]利用BRET技术检测PPAR-RXR异源二聚体的形成。Powell和Xu[56]利用BRET技术证明植物雌激素可选择性诱导ERα同源二聚体、ERβ同源二聚体和ERα-ERβ异源二聚体的产生,由于不同二聚体的功能差异显著,使得该方法可用于ER二聚体选择性的雌激素类物质筛选。Grossmann等[75]利用BRET研究MR同源二聚过程,发现二聚化发生在热休克蛋白(heat shock proteins, HSPs)解离以及核转移之后。Giner等[76]优化了BRET方法,可直接测定核受体二聚体对COA的募集,通过比较了维甲酸类物质(rexinoids)诱导的RXR-RXR同源二聚体、Nur77-RXR异源二聚体和Nurr1-RXR异源二聚体对COA的募集发现不同的同源和异源二聚体显示出对COA的差异性募集活性。Cotnoir-White等[77]在BRET的基础上开发了一种在活细胞中检测三元复合物的方法,即生物发光共振能量转移与荧光增强的联合转移(bioluminescence resonance energy transfer with fluorescence enhancement by combined transfer, BRETFect),并用该方检测E2、OHT等配体诱导的ERα-ERα同源二聚体与ERα-ERβ异源二聚体对COA的募集。
BRET不需要激发源,利用化学供体底物,背景荧光值可忽略不计,避免了FRET可能产生的光漂白问题,减少假阴性结果[56]。但BRET需要特定设备,且荧光素及底物价格昂贵,无法对比较弱的核受体间蛋白相互作用进行检测[73,78]。
3.3 双分子荧光互补(BiFC)
BiFC是最通用的蛋白质互补分析(protein-fragment complementation assays, PCA)技术,其原理是将FP切开形成不发荧光的2个片段(N末端和C末端),这2个片段分别与目标蛋白融合,如果目标蛋白相互作用,则2个互补片段彼此靠得足够近可恢复它们的荧光活性[78](图3(c))。
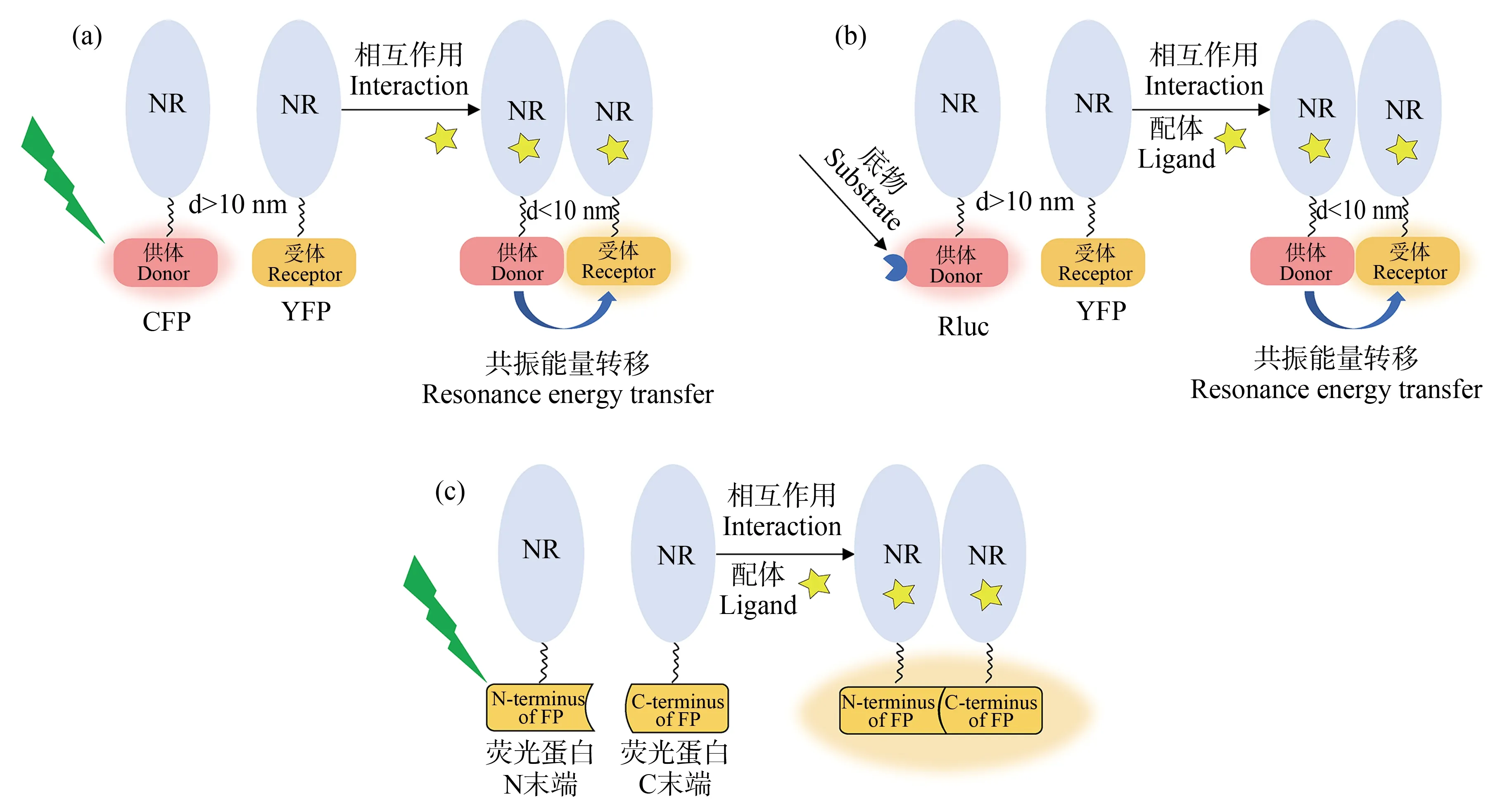
图3 FRET(a)、BRET(b)和BiFC(c)原理图注:FRET中当荧光供体蛋白和荧光受体蛋白间距离<10 nm时,供体将能量转移给受体;BRET利用生物发光酶作为供体分子, 荧光蛋白作为受体分子,当两者之间距离<10 nm时,供体将能量转移给受体;BiFC将荧光蛋白切开形成不发荧光的2个片段 (N末端和C末端),并分别与目标蛋白融合,如果目标蛋白相互作用,则会恢复荧光活性。Fig. 3 Schematic diagram of FRET (a), BRET (b) and BiFC (c)Note: FRET indicates that when the distance between the fluorescent donor protein and the fluorescent receptor protein is less than 10 nm, the donor transfers energy to the receptor; BRET uses bioluminescent enzymes as donor and fluorescent protein as receptor; when the distance between them is less than 10 nm, the donor transfers energy to the receptor; BiFC cleaves the fluorescent protein into two fragments (N-terminal and C-terminal) that do not fluoresce, which are fused with the target protein respectively; if the target protein interacts, the fluorescence activity will be restored.
利用BiFC将全长的ERα分别融合到YFP突变体(citrine-YFP)的N末端和C末端并共转染于COS-7细胞,发现ERα激动剂E2、BPA、染料木黄酮和对羟基苯甲酸丁酯(butylparabene),拮抗剂氟维司群(ICI)以及选择性雌激素受体调节剂4-羟基他莫昔芬(4-OHT)都能剂量依赖性地增加ERα-ERα同源二聚体荧光信号;有趣的是,ICI和4-OHT诱导的荧光信号高于E2,这可能是由于配体诱导的二聚体或构象差异所致[79]。雄激素受体剪接变异体(AR splice variants, AR-Vs)的表达上升被认为是产生以AR为靶点的药物抗药性的重要机制,Xu等[80]使用BiFC研究了2个主要的AR-Vs,发现它们不仅彼此间发生同源和异源二聚化,而且还与全长雄激素受体异源二聚化,阐明了AR-V介导基因调控的机制,并为合理的药物设计提供了重要的途径。Bedi[81]将LXRα贡献二聚化界面的关键氨基酸进行突变,使用BiFC表征LXRα突变体与RXRα和PPARα在活细胞中形成异源二聚体的能力,证明二聚化界面关键氨基酸突变能够削弱二聚化效应。
BiFC方法简单直观,既可以检测蛋白之间的相互作用,也可以定位相互作用蛋白质的位点,避免了用外源试剂处理细胞可能产生的问题[82]。但其目标蛋白容易受到FP片段的影响,当2个目标蛋白包含在单一复合物中但彼此没有相互作用时易产生假阳性结果[83]。
3.4 计算机模拟
随着计算机技术的发展,分子动力学模拟(molecular dynamics, MD)越来越多地被用来研究生物大分子作用[84]。但大多数MD模拟针对核受体单体进行研究,主要涉及配受体结合过程和共因子招募过程[51, 85],仅有少部分针对核受体二聚化过程进行模拟研究。
Zhuang等[86]利用经典的分子动力学模拟(MD)和随机加速分子动力学(random acceleration molecular dynamics, RAMD)模拟分别研究甲状腺激素T3与TRα-LBD、TRβ-LBD和TRα/LBD-RXR/LBD异源二聚体的解离效应,发现相比于TR单体,TR异源二聚体显著影响T3的解离,这为研究其他TR配体提供了重要信息。Sonoda等[87]利用部分增强采样分子动力学模拟(locally enhanced sampling molecular dynamics simulations)分别研究E2和雌激素受体调节剂雷洛昔芬(raloxifene, RAL)与ERα-LBD单体和ERα/LBD-ERα/LBD同源二聚体间的解离机制,发现相比于ER单体,其二聚化强烈抑制E2并改变RAL的解离路径。模拟研究中配体的解离路径的差异表明二聚化对于配体调节具有重要作用。Chakraborty等[88]利用分子动力学模拟证明ERα-ERα同源二聚体比ERα-ERβ异源二聚体更稳定。此外,Fratev等[89]利用加速分子动力学(accelerated molecular dynamics, aMD)模拟研究发现内分泌干扰物配体可以通过二聚体间的相互作用控制H12的位置,这对于核受体的激活至关重要。计算机模拟方法因其快速且省时省力等优点,已成为辅助内分泌干扰物质干扰核受体研究的重要手段之一。
4 总结与展望(Conclusions and prospect)
天然激素调控的核受体作为转录调控因子,几乎参与了人体所有组织和器官的功能调节。作为环境化合物的重要干扰靶点,当EDCs作用于核受体后,会导致人体内分泌系统紊乱,最终产生不良结局。EDCs主要通过模仿或拮抗天然激素,与核受体结合并影响核受体的二聚化过程,核受体通过同源或异源二聚后形成多聚体,进一步结合到DNA反应原件,通过共调节因子的招募过程最终调控转录活性。在以往研究中,人们主要着眼于受体-配体竞争结合、共调节因子招募以及核受体-DNA结合过程,但却无法完美解释许多内分泌干扰过程。近几年,越来越多的研究发现,核受体二聚化过程也对EDCs的内分泌干扰活性存在决定性的作用。因此,本文就EDCs介导的核受体二聚化转录机制、核受体二聚化与转录活性间的关系以及基于活细胞的核受体二聚化研究方法进行了概述,以期为深入理解内分泌干扰物质的分子机制,推进化合物内分泌干扰风险评估提供参考。
核受体二聚化过程对于研究化合物的内分泌干扰效应至关重要,已有进展主要针对EDCs对ER二聚化的影响,而关于EDCs与其他核受体二聚化过程的研究较少,探究EDCs对核受体二聚化的影响道阻且长。核受体二聚化机制复杂,环境领域研究核受体二聚化的过程极其依赖于生命科学技术的发展,但目前生命科学领域对二聚化特别是异源二聚化的作用机制和生物学功能尚不明确。解析核受体二聚化机制涉及很多生命科学相关实验,这对环境领域的研究人员而言困难重重,今后在探究EDCs对核受体二聚化影响机制的过程中需加强与生命科学的合作。
然而面对数以万计的未知内分泌干扰活性的环境污染物,生化实验已经无法对其内分泌干扰活性逐一检测,且利用生化实验进行二聚化测定费时费力费财。随着计算机技术的发展,分子动力学模拟、分子对接等相关技术越来越多地被应用于生物大分子作用的研究中。已有很多计算机辅助的相关技术被用于研究配受体结合和共因子招募过程,但有关内分泌干扰物对核受体二聚化影响的计算机模拟研究相对较少。今后可更多的将计算机技术应用于核受体二聚化过程的研究,并结合配受体结合和共因子招募过程,开发基于分子启动事件的核受体内分泌干扰筛查模型,为绿色化学的开发夯实基础。
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
华人时刊(2023年1期)2023-03-14 06:43:36
海洋通报(2022年4期)2022-10-10 07:40:26
农业工程学报(2022年5期)2022-06-22 12:15:58
汉字汉语研究(2021年2期)2021-08-30 08:58:46
中华戏曲(2020年2期)2020-02-12 05:17:58
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
河北书画研究(2016年3期)2016-04-28 08:55:35
合成化学(2015年4期)2016-01-17 09:01:11
华东理工大学学报(自然科学版)(2015年3期)2015-11-07 09:17:36
