
基于CRISPR/Cas9的毛果杨bHLH106转录因子的功能研究
2021-11-30 02:14孙佳彤国艳娇周晨光姜立泉
南京林业大学学报(自然科学版) 2021年6期
孙佳彤,国艳娇,李 爽,周晨光,姜立泉,李 伟
(林木遗传育种国家重点实验室(东北林业大学),黑龙江 哈尔滨 150040)
木材形成机制研究一直是林木研究的重点。木材的形成是一个高度有序且连续的过程,包括维管形成层的增殖、木质部细胞的分化扩张、次生细胞壁的沉积增厚、细胞的程序性死亡[1-2]。次生木质部包括纤维细胞、导管细胞和射线细胞[3]。已经从模式植物中鉴定出许多参与次生木质部发育的转录因子,它们形成转录调控网络用以调控木材的形成过程[4-6]。因此,探究转录因子在木材形成过程中的作用机制,可为利用林木分子生物学手段改良木材性状,提高木材质量提供理论依据。
bHLH(Basic Helix-Loop-Helix)是一类碱性螺旋-环-螺旋类转录调控因子,主要存在于真核生物中,是仅次于MYB的第2大转录因子家族。在植物中,bHLH转录因子家族对植物的调控发育、胁迫响应、信号传导和次生生长方面起着重要作用[7-8]。大多数bHLH蛋白是在模式植物拟南芥中被发现和鉴定的,其功能包括:参与调节信号传导合成代谢途径,如植物激素的合成、光信号传导[9-10]、光敏信号的调节[11-12]、脱落酸信号传导[13];参与植物的生长发育,如种子的萌发;调控植物抗逆胁迫,如低温胁迫[14]、盐胁迫[15]等。此外,bHLH基因家族同样参与木质部的生长发育,例如一个非典型的bHLH转录因子调节生长素下游的早期木质部发育[16];棉花 bHLH 蛋白GhFP1正向调节纤维细胞的伸长[17];在拟南芥次生细胞壁的相关转录组数据中,识别到了一些bHLH转录因子调控次生细胞壁的合成[18];在一项研究中确定一个bHLH转录因子二聚体TMO5/LHW是拟南芥中维管发育的关键调节因子[19],其缺失突变体中维管组织逐渐消失;bHLH转录因子LHW-T5L1二聚体促进木质部前体细胞中关键细胞分裂素基因的表达,导致周围细胞中细胞分裂素水平升高,从而促进原形成层细胞的特异性增殖[20]。然而,bHLH转录因子参与林木木材形成还鲜见报道。
基因编辑工具的出现,为基因功能的研究和植物性状的改良带来了很大的帮助[21]。CRISPR(clustered regularly interspaced short palindromic repeats)基因编辑技术在很大程度上提高了基因编辑的效率和实用性。目前,该技术已被广泛应用于基础科学、人类疾病治疗、作物基因功能及遗传育种等领域的研究中[22]。CRISPR/Cas9系统共包含了两个部分:向导RNA(sgRNA)和Cas9核酸酶,该系统的主要功能是在特定的基因组位点诱导DNA双链的断裂[23]。双链断裂之后非同源末端连接的细胞修复可能导致碱基的插入或缺失[24],从而改变碱基序列。利用CRISPR/Cas9技术可以对全基因组的任何一个基因进行基因编辑,可通过CRISPR/Cas9系统获得产量、品质和抗性都更高的优良品种[25]。例如通过CRISPR/Cas9技术编辑马铃薯R基因得到抗病马铃薯品系[26];以CRISPR 介导CCR的基因编辑技术获得了低木质素的新杨树优良品系[27]。自2015年首次将CRISPR/Cas9系统应用于杨树中以来[28],已有许多将CRISPR/Cas9系统应用于树木的基因功能研究。例如,利用CRISPR/Cas9技术创制毛果杨PtrVCS2基因突变体,通过对突变体植株的分析发现PtrVCS2促进形成层向木质部分化,使植株提前产生增厚的次生细胞壁,表明毛果杨PtrVCS2参与木材形成的调控[29];利用CRISPR/Cas9技术得到毛果杨形成层特异表达的PtrWOX4转录因子功能缺失突变体,使植株顶端优势被破坏,顶芽和根部发育不良,形成层发育受到显著影响,木质部分化紊乱[30]。
本研究通过分析东北林业大学林木遗传育种国家重点实验室姜立泉团队前期对野生型(WT)毛果杨茎干不同细胞类型进行的RNA-seq数据分析,鉴定出一个在形成层和次生木质部细胞中较高表达的PtrbHLH106基因,利用CRISPR/Cas9系统创制了毛果杨PtrbHLH106基因功能缺失突变体。通过对突变体的表型观察及分析,初步揭示该基因在木材形成方面的功能。
1 材料与方法
1.1 实验材料、载体及试剂
实验材料为东北林业大学林木遗传育种国家重点实验室(本实验室)保存的毛果杨植株,基因型为Nisqually-1。用次氯酸钠(NaClO)对野生型毛果杨侧枝进行消毒后,经植物组织培养进行芽诱导,获得无菌毛果杨组培苗。组织培养条件:温度25 ℃,光强约40 μmol/(m2· s),光周期为16 h光照/8 h黑暗。温室培养条件:温度范围22~25 ℃,光强约300 μmol/(m2· s),光周期为16 h光照/8 h黑暗。温室种植毛果杨使用135 ℃高温高压灭菌30 min后冷却的土壤。
pENTR载体(pENTR/D-topo)购自Life Invitrogen公司;T载体(pMD18-T)购自TaKaRa;pUC19-GFP载体为本实验室保存;pEgP237-2A-GFP由Keishi Osakabe实验室惠赠。RNA试剂盒(RNease plant Mini Kit)、胶回收试剂盒(GEL Extraction Kit)、PCR纯化试剂盒(PCR purification kit)、质粒提取试剂盒(Plasmid mini kit)均购自QIAGEN公司;反转录试剂盒(PrimeScript RT reagent Kit)购自TAKARA;Pfu 聚合酶(Pfu DNA Polymerase)购自Aglient公司;体外转录试剂盒(T7 quick high yield RNA synthesis kit)购自NEB;甲苯胺蓝染色剂购自Sigma;大肠杆菌(Escherichiacoli)TOP10感受态细胞、根癌农杆菌(Agrobacteriumtumefaciens)GV3101感受态细胞均为本实验室保存。
1.2 毛果杨PtrbHLH106基因的克隆与分析
根据PtrbHLH106基因的ID(Potri.006G037100),在毛果杨基因组网站(https://phytozome.jgi.doe.gov/pz/portal.html)下载基因序列,设计特异性引物(表1)。使用RNA提取试剂盒对毛果杨的分化木质部总RNA进行提取,使用反转录试剂盒进行反转录后得到的cDNA作为PCR扩增模板,对目的基因进行PCR扩增,PCR反应体系:10×Pfu Buffer 2 μL、dNTPs 2 μL、cDNA模板1 μL、正反向引物各1 μL、Pfu DNA Polymerase 0.4 μL、去离子水12.6 μL,反应体系为20 μL。PCR反应程序:94 ℃预变性2 min;94 ℃变性45 s,52 ℃退火30 s,68 ℃延伸30 s,30个循环;68 ℃再延伸30 min。PCR扩增获得该基因片段,并连接到pENTR载体上,经热激法大肠杆菌转化后,获得阳性克隆,测序检测PtrbHLH106序列。

表1 所用引物及序列
1.3 毛果杨PtrbHLH106基因gRNA选取及体外切割验证
使用在线工具(http://crispr.hzau.edu.cn/CRISPR2/)对PtrbHLH106进行gRNA的选取,选取gRNA的原则是分数高、脱靶概率低于0.5、GC含量在50%左右,且定位在外显子上。在挑选好的gRNA序列前加T7启动子(5′-TAATACGACTCACTATAGG-3′),后加stem-loop(5′-GTTTAAGAGCTATGCTGGAAACAGCATAGCAAGTTTAAATAAGG-3′),该序列作为gRNA的体外转录模板序列,用于gRNA的体外切割验证。
将gRNA体外转录模板序列经PCR反应生成双链DNA。PCR反应体系为:T7-bHLH106-stem-loop (0.5 μmol/L) 2 μL、BS6 (0.5 μmol/L) 2 μL、T25 (10 μmol/L) 5 μL、BS7 (10 μmol/L) 5 μL、dNTP Mix 1 μL、10×PCR Buffer 5 μL、rTaq1 μL、去离子水29 μL,共50 μL。PCR反应程序:95 ℃ 1 min;95 ℃ 10 s,59 ℃ 10 s,72 ℃ 10 s,45个循环;72 ℃,30 s。
PtrbHLH106基因gRNA的体外转录:利用体外转录试剂盒对gRNA进行体外转录,反应体系为:NTP Buffer Mix 7.5 μL、Nuclease-free water 3 μL、Template DNA 8 μL、T7 RNA Polymerase 1.5 μL,共20 μL。置于37 ℃恒温水浴锅中4 h。
模板载体的构建及模板质粒线性化:依据毛果杨网站提供的PtrbHLH106基因全基因组的序列信息及pUC19-35S-GFP载体上所含有的酶切位点,在gRNA前后共1 kb位置设计带有酶切位点的特异性引物(表1)。PCR模板为野生型的全基因组DNA,PCR扩增后对目标片段的胶回收产物和pUC19-35S-GFP质粒分别同时进行BamHⅠ和SalⅠ双酶切,T4 DNA连接酶进行连接,将连接产物利用热激法转化到TOP10大肠杆菌中,筛选阳性单克隆进行测序验证。测序正确的单克隆进行质粒的提取,使用PstⅠ进行模板质粒pUC19-PtrbHLH106的线性化,酶切产物纯化后获得线性化的体外切割模板DNA。
体外检测gRNA切割结果:检测体系为Cas9蛋白(1 mg/mL,本实验室前期纯化获得) 2 μL、gRNA 2 μL、线性化的DNA模板12 μL、Phosphate buffer (pH 7.5) 0.8 μL、DTT 0.02 μL、MgCl21 μL、PBS 2.18 μL。置于37 ℃恒温水浴锅1 h,1 h后迅速放入液氮中,随后用PCR纯化试剂盒进行产物纯化,经琼脂糖凝胶电泳检测并观察结果。
1.4 CRISPR基因编辑载体的构建
设计经体外验证的gRNA引物(表1),对gRNA进行引物复性。反应体系为:10×EX-TaqBuffer 2 μL、gRNA-F 9 μL、gRNA-R 9 μL,共20 μL。PCR反应程序如下:95 ℃ 30 s;72 ℃ 2 min;37 ℃ 2 min;25 ℃ 2 min。PCR产物稀释100倍待用。载体pEgP237-2A-GFP经BsaⅠ单酶切,与PtrbHLH106的gRNA用T4 DNA连接酶连接,将连接产物利用热激法转化到TOP10大肠杆菌中,筛选阳性单克隆进行测序验证,随后将阳性质粒pEgP237-U6-PtrbHLH106gRNA-35S-Cas9转入GV3101农杆菌中,用于毛果杨的遗传转化。
1.5 毛果杨遗传转化及转基因植株的鉴定
毛果杨遗传转化采用已建立的农杆菌介导法[31-32],选择生长30 d左右的健康毛果杨无菌组培苗,株高10~12 cm,切下第2~3节茎段,用农杆菌侵染20 min后放置暗处共培养48 h;暗培养结束后,用含有250 mg/L头孢霉素的无菌水浸泡清洗茎段,随后移至含有25 mg/L卡那霉素和250 mg/L头孢霉素的选择培养基上,培养25~30 d后,将抗性芽移至含有20 mg/L卡那霉素和125 mg/L头孢霉素的抗性芽生根培养基中。待植株生根后,剪下转基因植株的叶片并提取全基因组DNA,用pEgP237-2A-GFP上的F端引物M13F(表1)和PtrbHLH106基因gRNA的R端引物(表1)进行PCR扩增鉴定。
1.6 转基因植株PtrbHLH106基因编辑情况鉴定
参照毛果杨DNA序列,在包含gRNA位点的上下游共500 bp处设计引物,以转基因植株DNA为扩增模板,扩增目的片段DNA,加A尾后连接到T载体上,用热激法转化到TOP10大肠杆菌中,每个植株挑取30个阳性单克隆进行测序,与野生型序列进行比对。
1.7 毛果杨表型分析及茎的横切面解剖观察
分别在ptrbhlh106突变体和野生型毛果杨生长60、90和120 d时对其进行株高、地径测量,每个基因型取3株,重复3次测量。在ptrbhlh106突变体和野生型毛果杨生长120 d时,分别取第2、4、6、8、10茎节制作石蜡切片,利用甲苯胺蓝对切片进行染色,在M8数字扫描显微成像系统(Precipoint)下观察横切面细胞形态。
1.8 测序及数据分析
测序服务及全部引物的合成均由吉林省库美生物科技有限公司完成(www.comatebio.com)。
利用SPSS软件中的独立样本t检验法对各生长指标及细胞数进行差异显著性分析,当P< 0.05时,表示差异显著,当P< 0.01时差异极显著。用SigmaPlot 10.0软件进行制图。
2 结果与分析
2.1 PtrbHLH106基因的获得
研究团队前期通过激光显微切割技术(laser capture microdissection, LCM),分别精确收集了毛果杨野生型植株第8茎节的形成层、木质部和韧皮部细胞,对其进行RNA-seq数据分析,结果发现PtrbHLH106基因主要在形成层和木质部细胞中表达(图1),因此推测PtrbHLH106可能在形成层和木质部发育中发挥一定的调控作用。通过对PtrbHLH106的克隆及测序,获得723 bp的CDS序列。

标准化FPKM值即转录本丰度标准化为每千个碱基的转录每百万映射读取的片段数,误差线代表3个生物学重复的标准误。Normalized FPKM indicates normalized transcript abundances as fragments per kilobase of exon model per million mapped fragments.Error bars represent SE values of three independent experiments.图1 毛果杨PtrbHLH106基因在不同组织细胞类型中的表达Fig.1 The transcript abundance of PtrbHLH106 in different cell types in Populus trichocarpa
2.2 毛果杨PtrbHLH106基因突变体的创制
为了进一步研究PtrbHLH106在木材形成过程中的功能,采用CRISPR/Cas9基因编辑技术创制了毛果杨PtrbHLH106基因功能缺失突变体。在突变体创制前,利用体外切割技术对选取的gRNA进行了验证,以便确定gRNA的可用性。
2.2.1 gRNA的体外验证
通过gRNA网站预测,选取1条分数高、脱靶概率低、GC含量在50%左右的PtrbHLH106外显子上gRNA,切割的模板示意图如图2A所示,gRNA位于线性化DNA模板的-952 bp处。经过体外切割,琼脂糖凝胶电泳结果如图2B所示,在不加入gRNA或Cas9蛋白时,装有PtrbHLH106基因片段的模板DNA均未被切断(第1和2号泳道),在加入gRNA、Cas9蛋白和模板DNA后,DNA被有效切成2段,电泳条带在约4.5 kb和0.9 kb处(第3泳道),与理论长度相符。该结果说明,选用的gRNA序列可有效结合Cas9蛋白,并对靶位点进行切割,可用于后续实验。
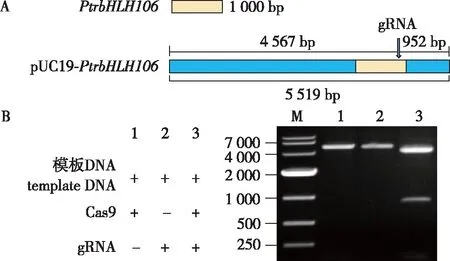
A. 模板DNA示意图schematic diagram of template DNA;B. 凝胶电泳结果the electrophoresis results(M. DNA marker DL10 000; 1、2. 对照组negative control; 3. 检测样品sample)。图2 gRNA体外验证Fig.2 The cleavage assay in vitro of gRNA
2.2.2 毛果杨遗传转化及转基因植株鉴定
构建CRISPR载体pEgP237-U6-PtrbHLH106gRNA-35S-Cas9,通过农杆菌侵染、抗性愈伤组织出芽、抗性芽在卡那霉素培养基上两次生根筛选过程(图3A—3C),获得3株抗性生根植株。提取待鉴定的抗性植株叶片基因组DNA,使用载体引物M13-F和PtrbHLH106的gRNA-R端引物进行PCR扩增,鉴定结果如图3D所示,两个阴性对照(第1和3泳道)均未扩增出片段条带,而抗性植株(第4~6泳道)均扩增出与阳性对照(第2泳道)相同大小的条带,说明PtrbHLH106重组质粒已经成功整合到毛果杨基因组中。

A. 愈伤组织分化诱导callus induction;B. 抗性芽诱导shoot induction;C. 抗性植株筛选screaning of the resistant plants;D. 转基因植株的PCR鉴定the PCR detection results(M. DNA marker DL 2000。 1. ddH2O为模板的阴性对照negative control with ddH2O as PCR template; 2. pEgP237-U6-PtrbHLH106 gRNA-35S-Cas9质粒为模板的阳性对照positive control with plasmid pEgP237-U6-PtrbHLH106 gRNA-35S-Cas9 as PCR template; 3. 野生型毛果杨叶片DNA为模板的阴性对照negative control with leaf DNA of wild-type plant as PCR template; 4~6. 抗性植株叶片DNA为模板的样品sample with leaf DNA of kanamycin-resistant plant as PCR template)。图3 毛果杨转基因植株的获得Fig.3 Generation of Populus trichocarpa transgenic plants
2.2.3PtrbHLH106基因编辑情况分析
对转基因植株的PtrbHLH106基因靶位点的编辑情况进行测序鉴定,结果如图4A所示:3个株系的突变体gRNA处均插入或缺失不同数量的碱基,如ptrbhlh106-1的两条等位基因分别插入1个碱基和缺失2个碱基,ptrbhlh106-2的两条等位基因分别插入1个碱基和缺失3个碱基,ptrbhlh106-3的两条等位基因分别插入1个碱基和缺失16个碱基。3个突变体植株均为双等位突变。编辑PtrbHLH106基因后的蛋白翻译情况如图4B,部分碱基的插入和缺失后,导致翻译提前终止,产生的氨基酸序列远短于正常的PtrbHLH106,ptrbhlh106-2的1条等位基因缺失了1个氨基酸同时造成1个氨基酸的改变,其他基因序列的翻译序列均未包含bHLH家族的典型HLH结构域[18]。该结果表明,已成功获得毛果杨ptrbhlh106突变体。为验证PtrbHLH106转录因子的功能,选取突变体植株ptrbhlh106-2、ptrbhlh106-3进行后续研究。
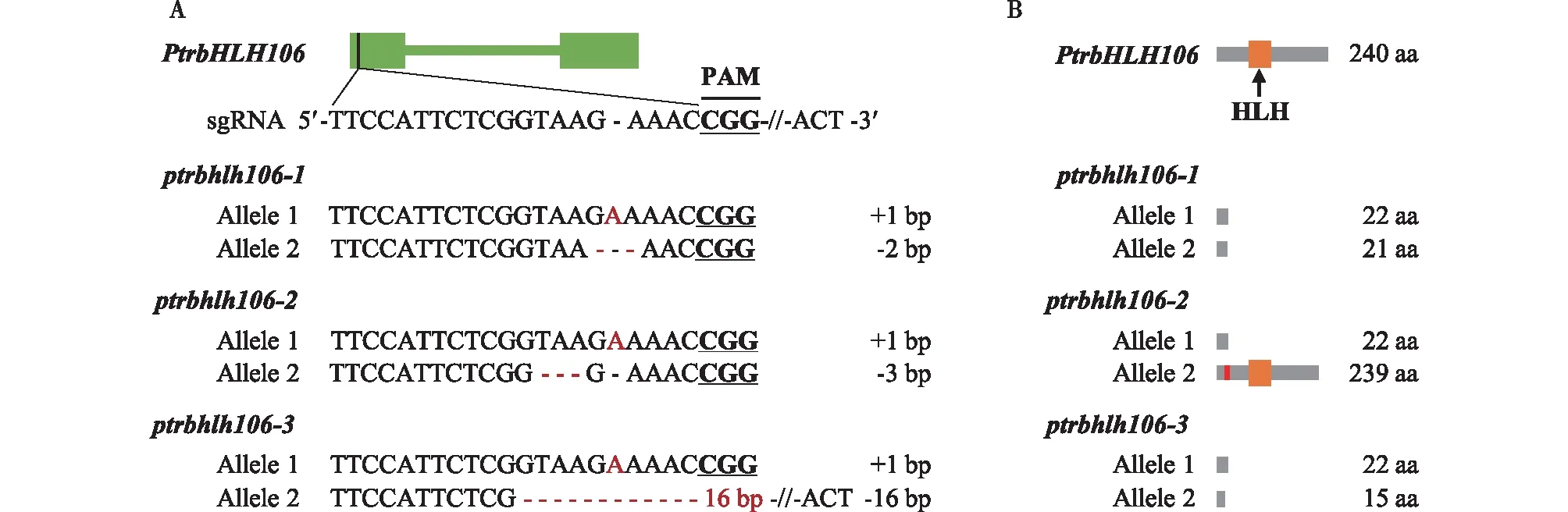
A. 等位基因编辑情况(红色字母表示碱基插入,红色“-”表示碱基缺失,“-16 bp-”表示缺失16个碱基)gene editing of alleles (The red letters represent insertion; the red short lines “-” represent deletion; “-16 bp-” represents 16 nucleotide deletion);B. 被编辑基因的蛋白翻译情况预测(红色块区域代表氨基酸改变) the prediction of amino acid sequence translation (The red short lines represent amino acid changed)。图4 毛果杨PtrbHLH106基因编辑情况Fig.4 Gene editing of PtrbHLH106 gene in Populus trichocarpa
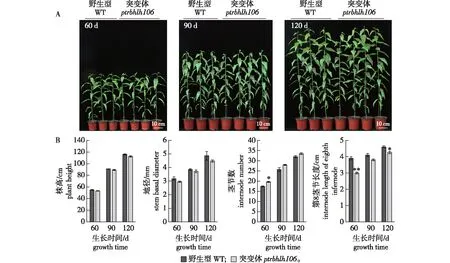
误差线代表由3个生物学重复计算的标准误,*表示通过t检验,突变体与野生型植物各株系之间存在显著差异,*.P<0.05,**.P<0.01)。下同。Statistical analysis of growth index of wild-type(WT) and ptrbhlh106 plants. Error bars represent SE values of three independent experiments. Asterisks indicate significant differences between each line of the mutants and wild-type plants by Student’s t test. The same below.图5 毛果杨野生型与ptrbhlh106突变体表型分析Fig.5 Phenotype observation of WT and ptrbhlh106 mutants in Populus trichocarpa
2.3 毛果杨ptrbhlh106突变体的表型分析
对ptrbhlh106突变体植株进行无性扩繁后,与高度相似的野生型毛果杨组培苗一起盆栽于温室中,在植株自然生长60、90和120 d时进行表型观察(图5)。从生长过程来看,毛果杨ptrbhlh106突变体与野生型相比,株高和地径没有明显差异。同时对茎节数以及第8茎节长度进行测定,统计结果表明,突变体植株在生长60 d时的茎节数量显著大于野生型,随着植株生长时间的延长,茎节数有增加的趋势,但差异不显著;第8茎节的长度在植株生长60和120 d时与WT差异显著,从植株生长周期来看,突变体植株第8茎节的长度有缩短的趋势。由以上结果初步推测,PtrbHLH106的功能缺失,对植株的生长未造成严重的影响。
为了进一步解析PtrbHLH106在杨树木材形成过程中发挥的功能,通过石蜡切片对毛果杨bhlh106突变体的第2、4、6、8和10茎节的横切面进行观察,结果显示突变体的各类型细胞的形态与野生型毛果杨无差异(图6A)。对切片中单位面积的导管细胞、纤维细胞数量及单个导管细胞的孔径进行统计分析,结果(图6C和6D)表明,与野生型相比突变体植株的纤维细胞数量显著减少,导管细胞数量无明显差异,单个导管孔径显著增加。对形成层细胞进行观察分析,发现突变体植株的形成层层数有增加的趋势,但差异不显著(图6B和6E)。以上结果表明,PtrbHLH106转录因子的功能缺失,导致木质部纤维细胞数量减少,导管孔径增大,初步说明该转录因子在次生木质部发育过程中发挥了调控作用。

A. 野生型(WT)和突变体(ptrbhlh106)毛果杨各茎节细胞形态观察(比例尺为200 μm)morphologic observation of stem internodes of WT and ptrbhlh106 (Bars=200 μm);B. 形成层细胞形态观察(红色线表示形成层区域,比例尺为100 μm)morphologic observation of cambium cells (Red line showed cambium areas, Bars=100 μm);C. 单位面积细胞数目统计statistics analysis of number of fiber and vessel cells in per unit cells;D. 导管孔径面积统计statistics analysis of lumen area of per vessel;E. 形成层细胞层数统计statistics analysis of number of cambium cell layer。图6 毛果杨ptrbhh106突变体石蜡切片观察及细胞统计Fig.6 Paraffin section observation and statistical analyses of cells in ptrbhlh106 in Populus trichocarpa
3 讨 论
因树木生长周期长、遗传背景复杂、基因组信息不完善等因素的制约,对树木的生长发育和环境适应等方面的研究受到很多技术的限制,尤其是在基因功能研究过程中,功能缺失突变体的创制异常困难。然而CRISPR/Cas9基因编辑技术在2015年首次用于杨树[28],就预示着树木突变体的创制不再是困扰科研人员的技术问题。应用CRISPR/Cas9系统成功获得了一个毛果杨转录因子的功能缺失突变体,为研究该转录因子功能提供了重要的遗传材料。在研究中,为了更加高效地获得毛果杨突变体,应用CRISPR/Cas9系统原理,开发了简易的体外切割实验,对选取的gRNA进行验证。实验结果也表明,经过验证的gRNA在毛果杨体内同样发挥着靶向高效性。该实验体系具有简便、耗时短、结果易观察等优点,但也存在DNA模板单一的缺点,后续将对该体系进行进一步优化。
不同的载体系统随着CRISPR系统在作物中的广泛应用应运而生,本研究使用的载体系统来自Osakabe研究团队[33-34],该载体系统可成功应用于苹果、葡萄等木本植物中,也同样成功地应用到林木中[32, 35]。本研究中获得的3株转基因毛果杨经测序鉴定,均为双等位突变体,说明该载体系统在毛果杨中具有很高的编辑效率。目前获得树木的单突变体已不再困难,但是获得树木的双突变体或多突变体,仍是对开发高效CRISPR系统的挑战。树木的遗传背景复杂,生长周期长,获得完整、高质量基因注释的树种还较少,许多树种的遗传转化系统不完善,缺少针对树木开发的RNA聚合酶Ⅲ类启动子,都是制约树木多突变体创制的因素。此外,基因的精准编辑如单碱基编辑已广泛应用于农作物中[36],而在树木中还未建立完善的碱基编辑系统。Li等[37]首次在杨树中成功利用碱基编辑器对木质素合成酶基因4CL进行单碱基突变,预示着CRISPR/Cas系统也将在树木中持续发挥其巨大的潜力。
木材形成是一个高度复杂的发育过程,涉及多类转录因子家族的参与和调控。从生物解剖学来看,木材是由次生木质部增厚的次生细胞壁形成的,而有关次生细胞壁的调控网络更是涉及多种转录因子,如NAC、MYB家族转录因子,分别在木质部纤维细胞、导管细胞的发育过程中起到分子开关、次级调控因子的作用[38-39]。除此之外,其他家族转录因子在木材形成调控网络中也直接或间接地调控木材形成相关基因的表达[6]。bHLH家族转录因子参与木质部的发育过程[16-18],本研究的PtrbHLH106属于该家族成员,主要在毛果杨的形成层和木质部表达。CRISPR/Cas9基因编辑的突变体ptrbhlh106在生长上并未受到影响,并且形成层的细胞层数有增加的趋势但变化不显著,说明PtrbHLH106的功能缺失未对整个植株的生长和形成层细胞的分裂分化造成明显影响。值得注意的是,在选取突变体做后续实验时,其中1株突变体的基因编辑结果并未造成PtrbHLH106氨基酸序列的较大变化,推测还保留该基因的功能。而两个突变体的表型一致,综合两个突变体的基因编辑情况和表型结果,说明可能是由于基因存在功能冗余[32],单独敲除1个基因,往往不能对树木重要发育过程产生很大影响。这一推测将通过后续获得多突变体等手段进行验证。
本研究在木材发育相关基因的功能研究中发现,bHLH106基因在木材发育方面具有重要功能。毛果杨ptrbhlh106突变体与野生型植株相比,虽然苗高生长和地径在统计学上没有显著的差别,但解剖视野中纤维细胞数量显著减少、导管孔径显著增大,形成层细胞层数增加,这说明PtrbHLH106在树木的木质部细胞发育和木材形成过程中,对调控径向生长量可能有重要的作用。在木本植物中,有关木质部发育的转录调控,与木材生物质累积和木质资源的生物产品转化高度相关[40-41]。PtrbHLH106基因对木材细胞壁纤维细胞的数量、导管的孔径大小,以及形成层细胞层数的调控,可能对木质部的组分,如纤维素含量、木质素S/G比例、含量甚至结构产生影响,从而改变木材的理化性质及终产品的加工利用工艺和品质。这也是杨树木材材性遗传改良的重要发展方向和研究重点[42-43]。诚然,本研究虽然拓宽了bHLH转录因子的功能范围,但有关PtrbHLH106基因的更多功能及其分子调控机制还需要进一步深入探究。
猜你喜欢
植物研究(2021年2期)2021-02-26
林业科学(2020年10期)2020-11-30
西藏人文地理(2020年4期)2020-11-19
农业工程技术·综合版(2020年1期)2020-04-23
复旦学报(医学版)(2019年5期)2019-10-09
天津农业科学(2015年11期)2015-12-03
西北植物学报(2015年3期)2015-07-04
山东农业科学(2014年4期)2014-07-18
安徽农学通报(2014年7期)2014-04-29
中成药(2014年11期)2014-02-28
