
基于三种贝叶斯方法的结核病基因数据挖掘及生物信息学分析
2021-11-26 06:54:42吴佩望
工程数学学报 2021年5期
张 旭, 吴佩望, 乔 峰
(西南大学数学与统计学院,重庆 400715)
1 引言
到目前为止,结核病仍然是全世界人类发病和因病死亡的主要原因,全世界每年有超过1000 万人感染结核病,160 余万人因结核病而死亡,更是有四分之一的人口是潜伏性结核病患者.目前中国乃至全世界在结核病防治问题上依然存在着很大的困难,如结核病发现率低、病原筛查率低、确诊率低、致病细菌耐药率高等.
目前我国对结核病的诊断主要通过痰涂片和病原分离培养.而痰涂片采用的方法灵敏度和特异度都较低,且受实验室条件及实验人员的影响较大.病原体分离培养则耗时较长,快速生长菌也至少需要3 天才有结果,时间长者甚至达到数周[1].因此,利用数据分析方法探索结核病发病机制,为疾病防控提供理论依据,并促进了结核病感染的分子诊断及治疗技术的发展.
人类全血以及外周血单核细胞的基因组表达分析已被广泛应用于检测活动性肺结核患者的宿主转录反应,并识别用于诊断的潜在生物标记物.例如:针对本文所用到的数据集,Cai 等[2]应用全基因组转录微阵列分析方法测定了结核病患者和对照组外周血单核细胞基因的表达量,并通过qRT-PCR 方法验证C1q 表达,发现与健康对照组和潜伏性结核感染患者相比,活动性结核病患者外周血中C1q 表达显著增加,证明了C1q 表达与人类结核中的活动性疾病相关,可能是区分活动性与潜伏性结核病以及结核性胸膜炎与非结核性胸膜炎的潜在诊断标志物.Blankley 等[3]对活动性结核病与健康对照组之间进行Benjamini Hochberg 多重检验校正的独立t检验鉴别了380 个差异表达的基因.Alam 等[4]利用Limma 包在健康组和活动性结核病患者之间鉴定出了266 个差异表达基因,其中149 个上调,117 个下调;在活动性结核病和潜在感染组之间共发现127 个上调基因和69 个下调基因.
然而,生命现象的复杂性与不确定性使得传统统计方法的使用存在很大的局限性,经典统计学方法一系列严格的前提假设并不能被很好的满足.比如,t检验往往需要在数据服从正态分布的假设下使用,但基因数据多数都不满足该假设,盲目使用将会使得研究结果与实际情况相去甚远.对于数据分析结果的验证目前也是一个棘手的问题.前面的这几篇文献对于选出的基因有的并没有做进一步验证,有的采用了qRT-PCR 方法验证,而该方法依赖于实验,耗时长、费用高,一般只能对极少数基因进行验证.因此,为了更加准确地模拟及预测具体的生物学过程,迫切需要一套基于数学、统计学以及计算机科学等的完整有效的组合方法,从海量的数据资源入手挖掘出隐含的、有价值的信息,与微生物学领域的研究方法形成互补,这也正是本研究的主旨.
本文先通过两种基于贝叶斯统计框架的方法,即线性模型及经验贝叶斯方法和信息先验性贝叶斯检验方法相结合,筛选出了结核病的潜在易感基因.再经过朴素贝叶斯分类器验证了这些易感基因的准确性,突出了贝叶斯方法在基因数据分析中的重要性.然后对这些基因进行了生物信息学分析,从生物学角度分析了结核病发病的分子机制.以期为结核病的诊断、长期防控工作提供参考依据.
2 数据和方法
2.1 数据来源及处理
本文所使用的数据集是来自美国国立生物技术信息中心(National Center for Biotechnology Information, NCBI)的高通量芯片表达谱数据库中的GSE54992,其中包含了9 例结核病患者样本(TB),12 例结核病潜伏患者样本和6 例正常样本(HD),每个样本有22283 个探针.该数据集应用全基因组转录微阵列分析方法测定了结核病患者和对照组外周血单核细胞基因的表达量[2].
本文还选取了NCBI 的另一组数据,同样来自NCBI 的高通量芯片表达谱数据库中的GSE83456 用作差异表达基因的验证,其中包含了45 例结核病患者样本(PTB)与61 例正常样本(HC),每个样本有47231 个探针.该数据集应用全基因组转录微阵列分析方法测定了结核病患者和对照组外周血单核细胞基因的表达量[3].
这两个数据集均已经过标准化,可以直接用于分析.在将探针转化为基因符号时,我们将对应同一基因符号的多个探针中的表达量的最大值作为该基因的表达量.
2.2 研究方法
2.2.1 线性模型及经验贝叶斯方法
线性模型及经验贝叶斯方法(Limma)是由Smyth 根据文献[5]提出的层次模型发展而来的,针对基因组微阵列数据“大p小n”问题的解决方法[6].Limma 整合了多种统计原理,它在基因表达值矩阵上运行,矩阵中每一行代表一个基因或者探针,每一列对应一个样本.一方面,通过加权或广义最小二乘法拟合多个线性模型,并以各种方式利用这些模型的灵活性.另一方面,由于基因组数据的高度平行性,它在每一个基因模型之间借用信息,允许基因之间及样本之间存在不同程度的差异,这使得统计结论在样本量较小的数据集中更为可靠.Limma 证明可以使用经验Bayes 后验方差估计进行精确的小样本推断.这种方法在小样本的实验中被证明是特别有优势的,确保了即使在重复次数很少的情况下,也能得到可靠和稳定的统计推断.Limma 是选取差异基因的一种基础的,传统的方法,该方法虽然缓解了由于样本量小而导致的推理结果不佳的问题,但是在估计低方差或高方差的基因时会引入偏差.
2.2.2 信息先验性贝叶斯检验
信息先验性贝叶斯检验(IPBT)是由Li 等[7]于2015 提出的一种替代方法,它在某种意义上与基于贝叶斯层次模型的Limma 方法“垂直”.Limma 方法中使用了经验贝叶斯方法借用同一次实验中不同基因的信息,而IPBT 方法则是借用在过去不同实验中相同的基因(或探针),但是使用相同的技术,相同类型的芯片,在同一类型的细胞上的测量信息.该方法的关键思想是基于丰富的历史数据为每个基因(或探针)指定了特异的信息性先验分布然后再进行贝叶斯假设检验.因为不同的基因具有不同的生物学功能,所以通常情况下它们的表达量会显示出相当多样化的分布情况.IPBT 的模型为
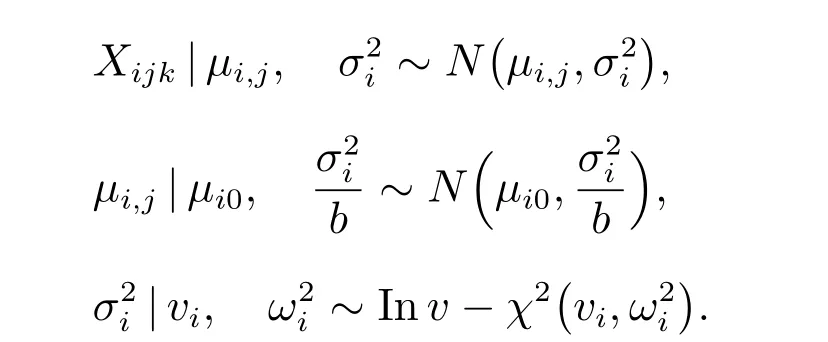

因此,与Limma 在同一实验中借用不同基因的策略不同,IPBT 假设每个基因都有属于自己独特的先验分布,然后在一次实验中借用这些历史数据中的先验信息来分析.经过与仿真数据的分析对比并绘制ROC 曲线,这种方法被证明筛选结果优于SAM、t检验及Limma 等其他方法[8].
2.2.3 朴素贝叶斯分类器
朴素贝叶斯是一种构造分类器的简单技术,是在贝叶斯算法的基础上进行相应的简化,即假设给定目标值时属性之间相互条件独立.也就是说没有哪个属性变量对于决策结果来说占有着较大或者较小的比重.虽然这个简化方式在一定程度上对贝叶斯分类算法的分类效果有稍许影响,但是在实际的应用场景中,极大地简化了贝叶斯方法的复杂性[8].朴素贝叶斯主要基于条件概率模型:给定一个要分类的问题,用向量x= (x1,x2,1,··· ,xn)代表n个特征,对于K个可能的类别中的每一类Ck,使用贝叶斯定理,将每一类的概率写为
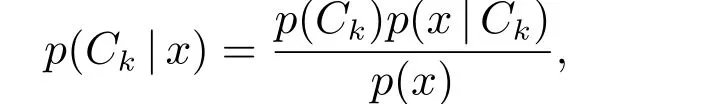
其中Ck类变量的条件分布是
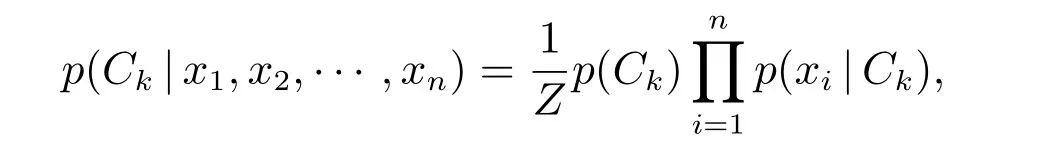
Z是一个只依赖于特征x的缩放因子,当特征变量的值已知时是一个常数.然后根据建立的朴素贝叶斯概率模型和最大后验概率决策准则构造朴素贝叶斯分类器[9]
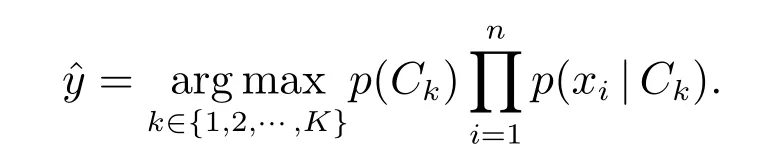
朴素贝叶斯分类器具有稳定的分类效率,在小规模数据集上的表现很好,能够处理多分类任务,并且对于缺失数据不敏感,在理论上朴素贝叶斯分类器与其他分类方法相比具有最小的误差率.
2.2.4 生物信息学分析
Gene Ontology(简称GO)是一个国际标准化的基因功能分类体系,是一项重要的生物信息学计划.而GO 富集分析是一种利用基因本体论分类系统解释基因组的技术,其中基因根据其功能特征被分配给一组预定义的领域,如细胞组分、分子功能、生物过程[10].本文中,我们利用文献[11]提供的R 语言包clusterProfiler 中的enrichGO 函数进行了GO 功能富集分析,并将其结果可视化.对差异表达基因进行生物信息学分析的另一种方案是KEGG 数据库[12],其中我们将根据基因所涉及的通路进行分组,并通过假设检验分析这些差异表达基因是否在某些通路上存在富集.本文中我们利用文献[11]提供的R 语言包clusterProfiler 中的enrichKEGG 函数进行了KEGG 通路分析.
3 结果与分析
3.1 筛选结果
GSE54992 数据集经过合并探针后,我们共得到13516 个基因的表达量数据.本文仅选取了其中的结核病患者和正常人两种样本数据进行对比分析.经过绘制PCA 图,如图1 所示,可以看出两组样本被明显分开,使我们有可能通过数据对比寻找差异基因.
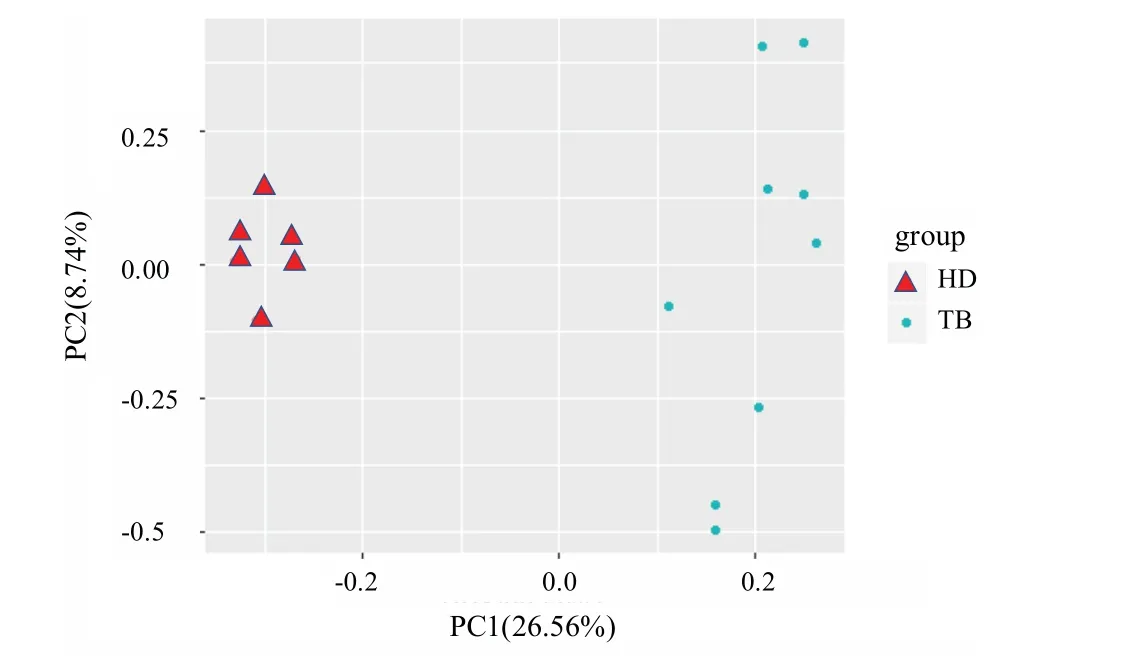
图1 GES54992 两组样本的PCA 图,HD 为正常样本,TB 为患病样本
我们先使用R 语言中的Limma 包处理基因表达量数据,一共得到558 个差异表达基因,其中279 个为上调表达基因,另外279 个为下调表达基因.接着我们又采用了信息先验性贝叶斯方法再一次筛选差异基因.基于文献[7]提供的GitHub 中下载得到IPBT 包,我们一共得到821 个差异表达基因,其中上调基因293 个,下调基因528 个.
为了使筛选的差异表达基因结果更为准确,我们对两种方法筛选出的差异表达基因取交集,得到了319 个基因,包括114 个上调基因和205 个下调基因.为了能更直观地看出结果,我们进行了可视化,绘制了这319 个基因的韦恩图和热图,如图2 和图3 所示.
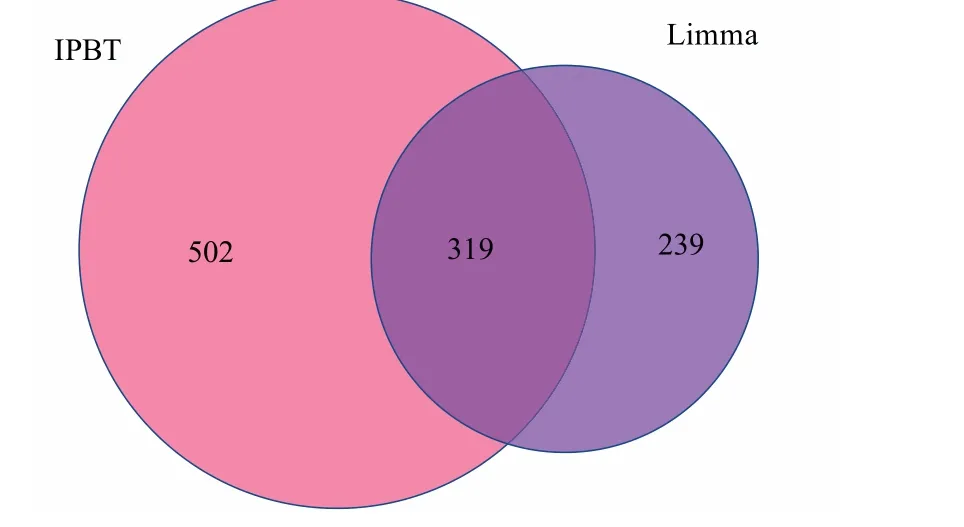
图2 IPBT 与Limma 筛选出的差异表达基因数量韦恩图,其中319 个基因被两种方法均识别

图3 319 个基因及15 个样本的表达热图
图3 中横轴是根据患病情况分组的15 个样本,纵轴是选出的319 个基因,每个小方块为一个基因的表达量.颜色的深浅代表该基因表达量的高低,红色越深表明基因表达量越高,蓝色越深表示基因表达量越低.从图中我们可以明显看出样本被分为两个组别,而上调与下调基因之间表达量也存在着很大的差异.表明我们通过这两种方法筛选出的差异表达基因的效果较好.
3.2 模型验证
由于数据集GSE54992 的样本本身的区分度较好(图1),我们选取了另一组数据GSE83456 来验证差异基因的分类效果.从图4 可以看出该数据集的两种样本有很大部分相互交叠在一起,难以区分开.
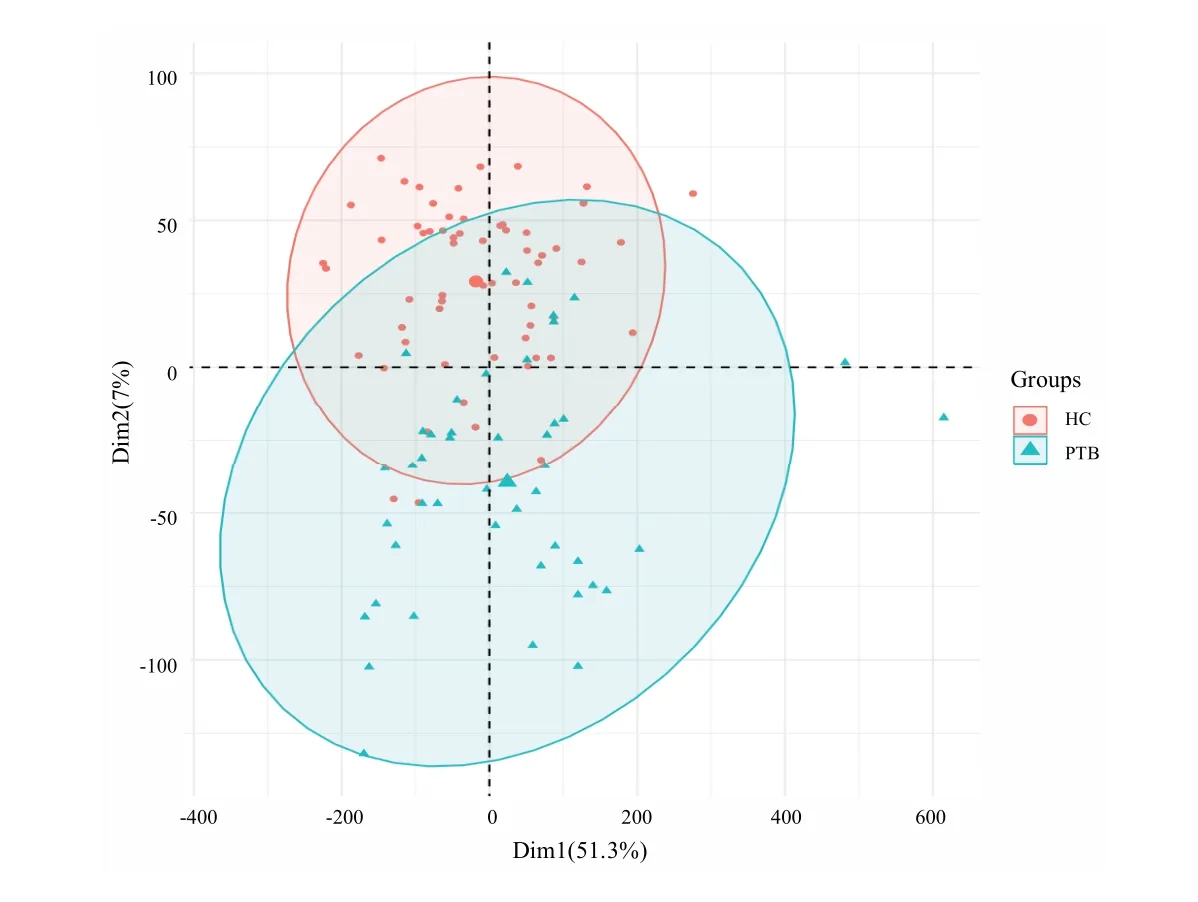
图4 GSE83456 两组样本的PCA 图,其中HC 为正常样本,PTB 为患病样本
对于GSE83456 这个独立验证集,我们采用两种方式选取了特征基因建立朴素贝叶斯分类模型进行对比,其一为IPBT 与Limma 两种方法共同筛选得到的319 个差异表达基因,其二为随机选取319 个基因.在70%训练集,30%验证集的条件下建立朴素贝叶斯分类模型,通过ROC 曲线我们发现,随机选取特征基因用作建模的准确率仅为55.6%,而通过基于贝叶斯方法筛选出来的差异表达基因用作特征基因对该数据集进行建模得到的分类准确率超过了85%,远远高于随机选取基因用作特征基因的表现.因此,我们认为筛选出的差异表达基因在结核病患病与否的预测上有较为显著的作用.
3.3 生物信息学分析结果
我们先将差异表达基因分为上调基因与下调基因,再分别进行了GO 功能富集分析,并根据p值大小分别选出上调和下调的各前十名,共20 项功能.经过文献查阅,我们发现选出的功能类别中有10 项功能与结核病发病相关,其中包括细胞迁移的正调节[13]、白细胞迁移[13]、髓样白细胞迁移[13]、细胞趋化性[14]、白细胞趋化性[14]、血管系统发育的调节[15]、血管生成[15]、运动的正调节[16]、细胞成分运动的正调节以及细胞运动的正调节[16].表明我们所筛选出来的差异表达基因中大多数基因被富集在与结核病发病相关的基因功能中.随后我们对选出的319 个差异表达基因进行KEGG 通路分析.由于无论是上调或是下调的基因均可能产生同一生物效应,或在同一通路中出现,因此,我们使用所有的差异表达基因一起进行KEGG 通路富集分析,通过查阅文献发现,p值列于前十的通路中,细胞因子-受体相互作用[17]、趋化因子信号通路[18]、抗生素的生物合成[19]与结核病发病相关,结核病与百日咳[20]、军团病[21]、阿米巴病[22]同属呼吸道疾病并且存在并发症的情况,更有文章表明用肿瘤坏死因子抑制剂治疗类风湿性关节炎可能会导致患活动性结核病的风险显著增加[23].此外,在所有p值小于0.05 的通路中,我们发现有9 个基因被富集在编号为hsa05152 的名称为“结核病”的通路中,这9 个基因分别为:IL1A、CAMK2A、IL1B、SPHK1、NFKB1、IL6、CR1、IL10 和JAK2.
4 结论与讨论
本文通过Limma 和IPBT 两种基于贝叶斯统计的方法从数据集GSE54992 中筛选出319 个差异表达基因;再基于独立验证集GSE83456 对筛选出的319 个差异表达基因利用朴素贝叶斯分类器进行验证,使原本难以分开的两组样本得到了较准确地分类,证明了筛选的差异表达基因的准确性,同时突出了贝叶斯方法在基因数据分析中的重要性;最后对这些差异表达基因进行GO 功能富集分析和KEGG 通路分析,找到了多个与结核病相关的功能与通路,使筛选出的差异基因得到了进一步验证.该研究对结核病生物标志物的筛选以及结核病的诊断与防控提供了新的思路.
本文巧妙地组合了三种贝叶斯方法,在传统的经验贝叶斯基础上结合了信息先验性贝叶斯检验来筛选差异基因,使得结果更加可靠;对选出的差异基因又利用朴素贝叶斯分类器进行验证,通过基因指导结核病样本分类的高准确率说明了选出基因的重要性.每个步骤都是基于贝叶斯框架,有利于减少系统误差.此外,IPBT 方法还有很大的提升空间,例如,目前IPBT 提供的历史数据仅有来自GPL96 平台的数十种细胞的微阵列数据,今后可以更多地收集相关历史数据以获得更准确的差异表达分析结果.
猜你喜欢
西部医学(2024年3期)2024-03-21 12:22:24
保健医苑(2022年5期)2022-06-10 07:46:46
电子测试(2018年1期)2018-04-18 11:52:35
数理化解题研究(2017年4期)2017-05-04 04:07:54
光学精密工程(2016年4期)2016-11-07 09:05:00
光学精密工程(2016年3期)2016-11-07 09:03:33
铁道通信信号(2016年6期)2016-06-01 12:10:20
电子器件(2015年5期)2015-12-29 08:43:15
中国卫生(2015年1期)2015-11-16 01:06:02
结核与肺部疾病杂志(2014年1期)2014-07-18 11:09:32
