
基于混酸回流制备碳点的中和过程
2021-11-22 01:20:26冯宁李洪光郝京诚
物理化学学报 2021年10期
冯宁 ,李洪光 ,郝京诚
1山东大学化学与化工学院,胶体与界面化学教育部重点实验室,济南 250100
2中国日用化学工业研究院,太原 030000
1 引言
碳点(carbon dot)一般是指尺寸在10 nm以下具有准球形结构,可稳定发光的纳米碳,因具有奇特的结构和优异的物理化学性质,尤其是光致发光特性,受到不同领域科学家的广泛关注1–6。碳点的制备可分为“自上而下”和“自下而上”两种方法,随着碳点研究的深入而被不断完善。碳点最早于2004年,Scrivens等人利用混酸回流提纯碳纳米管的过程中偶然发现7。2006年,Sun等人对一种经过特殊处理的碳粉进行激光刻蚀,并通过后续表面钝化,开启了碳点实验室制备的先河8。2008年,Giannelis等开始尝试利用小分子热解的方法制备碳点9,10。他们通过改变原料,获得了水溶性和油溶性两类碳点。目前,碳点的制备方法已经比较成熟,“自上而下”和“自下而上”两种方法均获得了长足的发展。前者除上述提到的混酸回流和激光刻蚀,还包括康振辉等人发展的电化学方法11。而后者的热解过程也被大大丰富,分为直接加热、水热、溶剂热、微波加热、超声等等。
作为最初发现碳点时所采用的制备方法,混酸回流在碳点研究中具有独特性。所选用的富碳前驱体,由最初的碳纳米管7,12,拓展到石墨棒11、石油焦13–15、煤16–19、头发20、碳纤维21、活性炭22以及各种炭灰23–26。混酸回流所制备的碳点,表面一般修饰有各种含氧有机官能团,尤其是羧基和羟基。反应结束后溶液为强酸性,使后续分离提纯过程,尤其是渗析操作难以进行。其次,碳点在高度酸化的反应液中被过度质子化,导致其水溶性大大降低。因此,反应液的中和是必需的步骤。中和过程中,所加入的碱性试剂不仅与溶液中过量的酸反应生成盐,碳点表面的羧基和部分羟基也将参与反应。我们近期的研究发现,由于碳点结构的复杂性,混酸回流制得的碳点,其表面酸性官能团的活性并不一致27。这一点,在其它表面富含羧基和羟基的纳米碳中也有体现,典型的如富勒醇28。这意味着中和过程中,这些酸性官能团反应的程度,将与所加入的碱性试剂的种类密切相关。同时,不同中和试剂的使用会引入不同的反离子,也将对碳点的性能产生影响。然而,以往的研究重点关注了富碳原材料、混酸比例、回流温度和时间对碳点性质的影响,而缺乏对中和过程的系统研究。这种针对具有同质内核不同表面化学属性碳点的系统研究,能够从另一个侧面,深入了解碳点的光致发光特性。
本文中,我们以富勒烯制备过程中产生的炭灰为原料,通过混酸回流制备碳点。采用多种技术手段,系统研究了NaOH、Na2CO3、K2CO3和NH3∙H2O四种不同中和试剂的运用对碳点结构和发光特性的影响。
2 实验部分
2.1 试剂
富勒烯炭灰购自苏州大德碳纳米科技有限公司,浓硫酸(H2SO4)和浓硝酸(HNO3)购自莱阳康德化学有限公司,氢氧化钠(NaOH)、碳酸钠(Na2CO3)、碳酸钾(K2CO3)购自莱阳康德化工有限公司,氨水(NH3∙H2O)购自天津凯美尔化学试剂有限公司,四乙基溴化铵(N(C2H5)4Br)购自阿拉丁,十四烷基二甲基氧化胺(C14DMAO,30% (w,质量分数)的水溶液)购自科莱恩公司(德国)。在整个实验过程中使用由UPH-IV超纯水净化器(中国)制备的电阻率为18.25 MΩ∙cm的超纯水。
C14DMAO水溶液冻干后,使用丙酮重结晶三次。其它试剂未经纯化,直接使用。
2.2 碳点的制备
将12 g富勒烯炭灰加入到100 mL浓硝酸和300 mL浓硫酸的混合液中,100 °C搅拌回流36 h。冷却至室温后,在冰水浴中加入超纯水稀释反应液,使反应液最终体积约为2 L。
将反应液等分为4份,向其中分别加入NaOH、Na2CO3、K2CO3和NH3∙H2O至溶液pH值约为7 (pH试纸监测)。过滤除去析出的无机盐,将滤液置入截留分子量为1000 Da的透析袋中,在纯净水中渗析1周左右,期间每隔2天换一次水。将渗析所得水溶液置于鼓风干燥箱中,70 °C下干燥备用。
在碳点的离子交换过程中,将过量N(C2H5)4Br加入NH3∙H2O制备所得的碳点水溶液,渗析4天,将渗析液冻干,得反离子为的碳点。
2.3 碳点与表面活性剂的复配
将C14DMAO配制成100 mmol∙L−1的母液,将碳点配制成0.5 mg∙L−1的母液。向一系列玻璃小瓶中,移入0.6 mL碳点母液和不同体积的C14DMAO母液,补加适量水使溶液最终体积为3 mL。计算得碳点的最终浓度为0.1 mg∙L−1。
2.4 测试与表征
使用DSC 822e (Piscataway,NJ)进行热重分析(TGA),测试条件为:氮气环境中,扫描速度为5 °C∙min−1;使用FTIR分光光度计(VERTEX-70)获得傅立叶变换红外(FTIR)光谱;元素分析使用有机元素分析仪(Vario EI III)得到结果;X射线光电子能谱仪(ESCALAB 250)用单色化AlKαX射线源(1486.71 eV)得到X射线光电子光谱(XPS);用紫外可见分光光度计(U-4100)获得紫外–可见光谱;使用荧光分光光度计(LS-55)获得荧光光谱,为便于对不同样品的荧光强度进行比较,测试在相同的仪器参数下(激发狭缝为2.5,发射狭缝为10)完成。
3 结果与讨论
3.1 碳点的基本结构
根据原材料和制备方法的不同,混酸回流所获得碳点的结构存在差异,但基本结构比较明晰。如图1a所示,中和前该类碳点包含一个碳质内核(carbonaceous core),其外围被各种含氧有机官能团(organic functional group)包裹。其中,羧基、羟基等酸性有机官能团因所连接碳原子杂化方式的不同,而具有不同的解离度。加入碱性试剂后,碳点外围的羧基和部分羟基如酚羟基将发生中和反应,所形成的碳点(图1b)可视作一种结构特殊、式量超大的盐,在空气中久置会发生潮解。

图1 碳点结构示意图。(a)中和前;(b)中和后Fig. 1 Illustration of the structure of the carbon dot.(a)before neutralization, (b)after neutralization.
本研究中,中和前的碳点来源于同一组反应,因此具有相同的结构。中和后碳点的结构和性质,将直接取决于所选用中和试剂的碱性及所含阳离子与碳点表面阴离子官能团的结合能力。下面,我们对NaOH、Na2CO3、K2CO3和NH3∙H2O四种试剂中和所得碳点的结构和性质进行详细剖析。
3.2 中和试剂对碳点结构的影响
图2a示出四种碳点的热重分析曲线。其中,100–200 °C之间的失重归因于碳点所含的结晶水。从图中可以看出,Na2CO3中和所得碳点结晶水含量最低,约为15% (w),而NH3∙H2O中和所得碳点结晶水含量最高,约为20% (w)。四种碳点200 °C之前失重曲线的斜率也存在差异。碳点结晶水含量的不同,一方面可归因于不同中和试剂所得碳点表面残存羟基数目的差异,另一方面则归因于所形成碳点(结构特殊的盐)与水分子的不同结合能力。
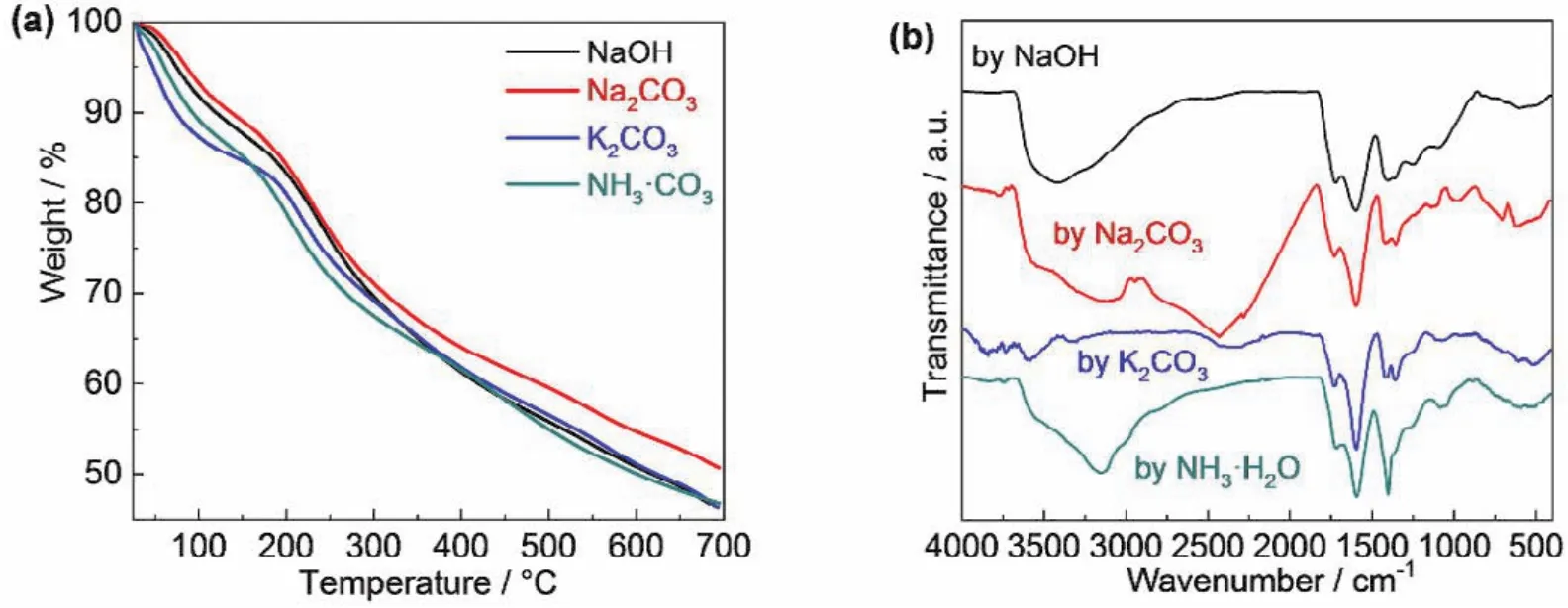
图2 四种碳点的(a)热重分析曲线和(b)傅立叶变换红外光谱Fig. 2 (a)TGA and (b)FTIR curves of the four carbon dots.
碳点表面羟基数目和所含结晶水的差异,在红外光谱中亦有明显体现。如图2b所示,四种碳点在3000–3400 cm−1范围的O-H伸缩振动表现出明显差异。从图中还可以看出四种碳点在1600 cm−1处的C=O伸缩振动,与图1所示的碳点结构一致。而1400 cm−1处C=C伸缩振动的存在则清晰地表明碳点碳质内核中含有sp2杂化的碳原子。
四种碳点的元素分析结果示于表1。可以看出,碳元素含量均在40% (w)左右。由于制备过程中使用了浓硫酸和浓硝酸,四种碳点均有少量氮、硫掺杂。其中,NH3∙H2O制得的碳点,由于外围铵根离子的引入,导致氮元素含量明显高于其它碳点。
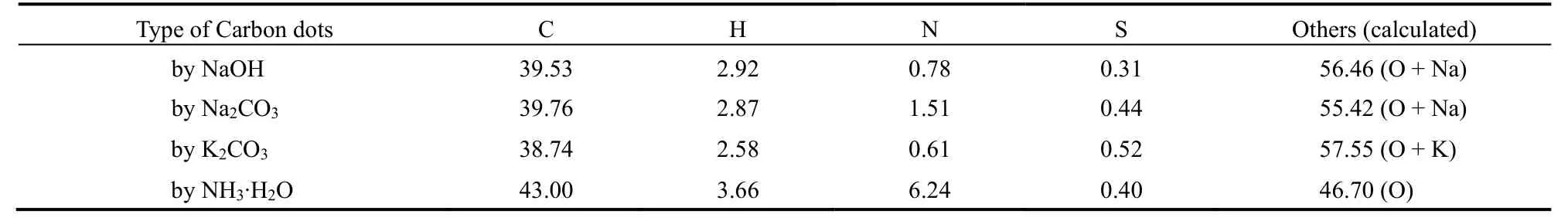
表1 四种碳点的元素分析结果(w/%)Table 1 Elemental analysis of the four carbon dots (w/%).
为了进一步获取碳点的结构信息,我们对四种碳点进行了X射线光电子能谱分析,结果示于图3。从全谱(图3a)中可以看出,四种碳点均表现出较强的C 1s和O 1s信号,表明碳点主要由碳、氧两种元素组成。NaOH和Na2CO3制得的碳点在1072 eV处有明显的Na 1s峰。K2CO3制得碳点的K 2p信号在全谱中被O 1s的信号峰掩盖,但在进一步高分辨谱图中,能够清晰地看到它在295和292 eV处的2p1/2峰和2p3/2峰(图3b)。氮元素因含量较低,其信号峰在全谱中不明显,只有NH3∙H2O制得的碳点在406–398 eV之间的N 1s峰能够被勉强辨认出来。而硫元素因含量太低,其信号峰在全谱中无法体现。但高分辨谱图显示四种碳点在172–165 eV的范围内,均出现了微弱的S 2p信号,且能够被拆分为S-C和S=O两类信号峰。作为一个典型示例,图3c给出了NaOH制得的碳点S 2p的高分辨图谱。

图3 四种碳点的X射线光电子能谱测试结果。(a)全谱;(b)K2CO3制得碳点的K 2p高分辨谱图;(c)NaOH制得碳点的S 2p高分辨谱图;(d–f)四种碳点C 1s (d)、O 1s (e)和N 1s (f)的高分辨谱图Fig. 3 XPS survey of the four carbon dots. (a)The spectra within the whole range; (b)High-resolution K 2p spectrum of the carbon dots prepared from K2CO3; (c)High-resolution S 2p spectrum of the carbon dots prepared from NaOH;(d–f)High-resolution C 1s (d), O 1s (e)and N 1s (f)spectra of the four carbon dots.
我们对四种碳点的高分辨C 1s、O 1s和N 1s谱图进行进一步分析,结果示于图3d,f。C 1s(图3d)可拆分为C=O、C-N/C-OH和C-C。三者的峰面积随中和试剂的改变而出现变化,表明中和试剂的确对碳点的表面有机官能团种类产生了影响。O 1s(图3e)谱图可拆分为O-C和O=C。与C 1s类似,二者的峰面积随中和试剂的改变出现变化。由于含量较低,N 1s的信噪比与C 1s和O 1s相比较差,但仍可拆分为N=O、N-H和N-C (图3f)。NH3∙H2O制得的碳点由于外围的引入,NH峰面积有一个明显提升。
如图1所示,与其它通过混酸回流得到的碳点一样,本文中由富勒烯炭灰为原料制备的碳点表面带有负电荷。我们曾对NaOH中和得来的碳点进行详细结构解析27,发现其表面电荷在45–55之间,表面电荷密度为3.6–4.4 nm−2。将其与两性离子表面活性剂十四烷基二甲基氧化胺(C14DMAO)复配时,碳点表面电荷随C14DMAO的持续加入而不断变化,使体系表现出丰富的相行为27。这一现象,为我们提供了一种对比研究不同中和试剂制备所得碳点表面化学属性的方法。
我们固定碳点的浓度为0.1 mg∙mL−1,C14DMAO的浓度在0.2–45 mmol∙L−1之间变化。四种复配体系的相图示于图4。C14DMAO在水溶液中会部分质子化,形成荷正电的聚集体。该聚集体与荷负电的碳点相互作用,导致澄清的溶液变浑浊。当复合物表面电荷过低,不足以稳定胶体体系时,溶液中产生沉淀。继续加入C14DMAO导致了沉淀的逐渐溶解。这一变化规律,与我们近期的研究结果一致27。在荷正电碳点/阴离子表面活性剂复配体系中,我们也观察到了类似的相行为29。
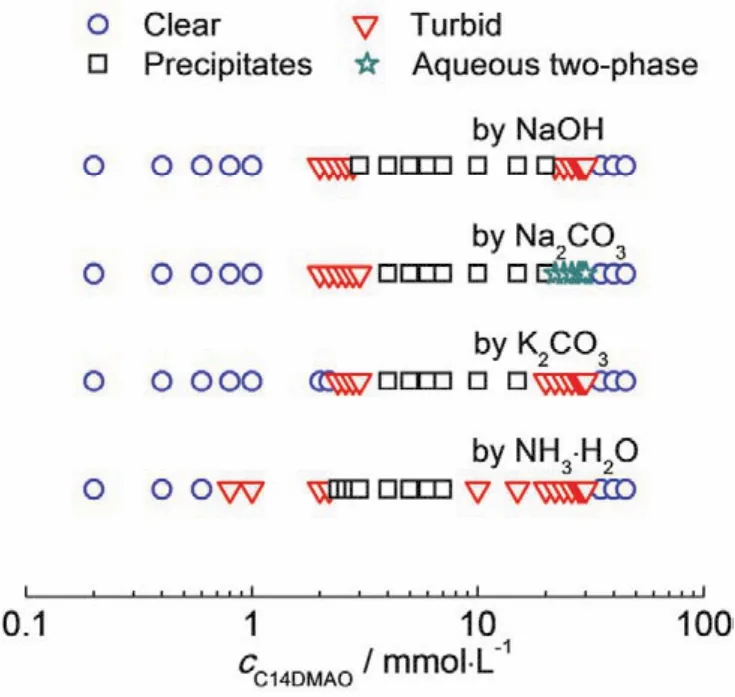
图4 四种碳点与C14DMAO复配后的相行为Fig. 4 Phase behavior of the carbon dots/C14DMAO.
从图4中可以看出,尽管溶液外观的变化规律一致,四种碳点表面化学属性的不同导致相分界所对应的C14DMAO的浓度存在差异。NaOH制得的碳点在C14DMAO浓度为2.0 mmol∙L−1时出现浑浊,在C14DMAO浓度为2.8 mmol∙L−1时出现沉淀。Na2CO3制得的碳点出现浑浊所对应的C14DMAO浓度与NaOH制得的碳点相同,但出现沉淀所对应的C14DMAO浓度略大,为3.0 mmol∙L−1;在沉淀区和澄清样品之间,并未形成均一的浑浊相,而是形成了一种上层浑浊,下层澄清的双水相。这种双水相与表面活性剂水溶液形成的双水相30–33相比,稳定性稍差,久置之后会转变为沉淀。K2CO3制得的碳点沉淀区对应的C14DMAO浓度范围与NaOH和Na2CO3制得的碳点相比略窄。
对于NH3∙H2O制得的碳点,当C14DMAO的浓度仅为0.8 mmol∙L−1时就已经开始出现浑浊;当C14DMAO的浓度为2.4 mmol∙L−1时,体系产生沉淀;其沉淀溶解所对应的C14DMAO浓度(7.0 mmol∙L−1)亦比其它碳点低(15.0或20.0 mmol∙L−1)。这可能是由于NH3∙H2O制得的碳点由于反离子空间位阻较大,导致解离度高,从而使碳点带有更多负电荷的缘故。
3.3 中和试剂对碳点光谱学特性的影响
图5示出了四种碳点水溶液(0.1 mg∙mL−1)的紫外-可见吸收光谱。从图中可以看出,NaOH、Na2CO3和K2CO3制得的碳点,曲线形状类似,随波长的增加,吸收强度逐渐降低,整个测试范围内未出现明显的吸收峰。NH3∙H2O制得的碳点,在331 nm附近出现了明显的吸收峰,可归因于碳点表面含氧官能团如与碳质核心中sp2杂化碳原子间的n–π跃迁。
我们对四种碳点的光致发光特性进行了详细研究,结果示于图6。从图中可以看出,四种碳点均表现出激发波长依赖性,随激发波长的增加,发射光谱红移。这与文献报道的众多碳点的光致发光特性一致1–6。四种碳点的最佳激发波长,均在460–470 nm之间。NaOH、Na2CO3和K2CO3制得的碳点,最佳发射波长在525 nm,而NH3∙H2O制得的碳点的最佳发射波长红移至533 nm。后者光致发光特性的与众不同,与其紫外–可见吸收光谱的结果保持一致。可以看出,使用NH3∙H2O这种弱碱对混酸回流的样品进行中和,与使用强碱如NaOH或强酸弱碱盐如Na2CO3和K2CO3相比,能够对碳点的光谱学特性进行更大程度地调节。为进一步了解产生这种差异的原因,我们利用离子交换的方法,将交换为四乙基铵根离子,研究发现,碳点的最佳发射光谱进一步红移至563 nm处(图7)。可见,除了中和过程中碳点表面酸性有机官能团反应程度的差异,反离子的不同也是导致碳点发光性能差异的重要因素。

图6 四种碳点水溶液(0.1 mg∙mL−1)的荧光光谱Fig. 6 Fluorescence of aqueous solutions of the four carbon dots (0.1 mg∙mL−1).

图7 反离子分别为碳点水溶液(0.1 mg∙mL−1)的荧光光谱Fig. 7 Fluorescence of the aqueous solution of the carbon dots (0.1 mg∙mL−1)with as the counterions, respectively.
从图6中还可以看出,四种碳点的荧光强度有着显著差异。四种碳点荧光强度的顺序为:Na2CO3> K2CO3> NaOH > NH3∙H2O。对比Na2CO3和NaOH做中和试剂,其差别在碱性不同,中和试剂的碱性过强对碳点的发光不利,而NH3∙H2O制得碳点较弱的发光特性表明中和试剂碱性太弱亦不利于碳点的发光;对比Na2CO3和K2CO3做中和试剂,其差别在于阳离子的不同,使用Na+作为反离子更有利于提高碳点的发光性能。
通常认为,碳点的发光主要来源于两个方面:一是碳质核心中π共轭基团,二是表面各种有机官能团形成的表面态34–40。本文中所研究的四种碳点来源于同一批混酸回流的样品,因此具有相同的碳质核心。它们光谱学特性的不同,主要归因于不同中和试剂的使用所导致的表面化学特性差异。这一结论,与四种碳点的结构表征结果相一致。然而,受制于碳点结构的复杂性,其发光机理目前仍不明朗,因此不同中和试剂对碳点光谱学特性的具体影响机制还需要进一步探究。尽管如此,我们的研究清楚地表明,对混酸回流所得的碳点,中和试剂的选用是影响其结构和性能的重要因素。
4 结论
混酸处理的过程中,中和作为重要的一环,中和试剂的选择至关重要。我们的研究结果表明,尽管中和过程对碳质核心影响很小,但不同中和试剂的运用将会对碳点的表面化学属性产生重要影响,并最终影响其光谱学特性。这一结果提示我们:除了继续寻找理想的富碳原材料、优化混酸回流工艺,中和试剂的合理运用也是优化碳点结构和性能的重要方面。
猜你喜欢
中学化学(2024年2期)2024-06-17 04:01:47
中国兽药杂志(2021年7期)2021-08-13 02:37:22
潍坊学院学报(2021年2期)2021-07-22 07:59:04
艺术品鉴(2020年5期)2020-07-27 02:43:08
中国电气工程学报(2019年27期)2019-10-21 10:25:52
中国有色冶金(2018年4期)2018-01-31 16:50:01
商情(2017年33期)2018-01-24 22:45:44
分析化学(2017年12期)2017-12-25 12:44:25
分析化学(2017年12期)2017-12-25 07:05:04
江苏理工学院学报(2017年2期)2017-07-09 21:02:05

- 物理化学学报的其它文章
- Photocrosslinking-Immobilized Polymer Vesicles for Lowering Temperature Triggered Drug Release
- Poly(ε-caprolactone)-Polypeptide Copolymer Micelles Enhance the Antibacterial Activities of Antibiotics
- ReaxFF MD局部区域反应追踪与物理性质可视化分析
- 稀土-天然皮革可穿戴X射线防护材料的合成及性能
- 石墨烯玻璃透明薄膜加热特性
- Single-Molecule Field-Effect Transistors with Graphene Electrodes and Covalent Pyrazine Linkers