
含氮金属有机框架衍生的铜基催化剂电催化还原二氧化碳
2021-11-22 07:01金惠东熊力堃张想连跃彬陈思陆永涛邓昭彭扬
物理化学学报 2021年11期
金惠东,熊力堃,张想,连跃彬,陈思,陆永涛,*,邓昭,彭扬,*
1苏州大学能源学院,能源与材料创新研究院,江苏 苏州 215006
2江苏省先进碳材料与可穿戴能源技术省重点实验室,江苏 苏州 215006
1 引言
人们对能源的需求随着人类社会和经济的发展而迅速增加。然而大规模利用化石能源导致了大量的二氧化碳排放,给地球造成了严重的温室效应,进而给环境带来了一系列问题。寻找如太阳能、风能、氢能等可再生能源来替代传统能源显得尤为迫切。然而这些可再生能源主要是以电能的形式得到利用,存在存储运输成本及损耗问题,而二氧化碳还原反应能够得到高附加值的能源和化学品,便于存储运输及使用,具有广阔的应用前景。人工还原二氧化碳的方法有几种,包括了光催化还原二氧化碳、光电催化还原二氧化碳、化学转化二氧化碳及电化学还原二氧化碳1等。其中,由于电化学还原二氧化碳(ECR-CO2)具有较高的能量效率和较为温和的反应条件,是当前还原二氧化碳最有前景的策略之一。然而由于线性二氧化碳分子超高的热力学稳定性,将二氧化碳分子活化为二氧化碳自由基阴离子(CO2•-)需要很大的能量(ESHE= -1.90 V)。与此同时,析氢反应(ESHE= -0.42 V)的能量则要小得多,因此整个反应过程都伴随着析氢反应(HER)2,导致产物选择性较低,因此寻找和开发高性能的电催化剂是解决这些问题的关键。
相对于CO,甲酸等常见的CO2还原产物,多碳化合物,如乙烯,乙醇和乙酸等,具有更高的能量的密度和商业价值。据研究表明,铜(Cu)是目前已知的唯一一种能单独将二氧化碳还原为高附加值产物的金属。最早报道铜作为ECR-CO2电催化剂的是Hori等3–5,他们发现在适当的负电位下,铜电极上能够生成甲烷(CH4)和乙烯(C2H4)等碳氢化合物。然而,Cu在电催化还原CO2过程中往往需要较高的过电势,这使得其发生严重的析氢反应,导致CO2整体催化活性较低,与此同时,由于Cu对大多数CO2还原产物的中间体吸附能都适中6–8,导致它对目标产物的选择性很低,可以同时生成许多还原产物,从两电子产物(如CO和甲酸9)到八电子产物(如甲烷)以及更高的多电子产物(如乙烯、乙醇)不等5,10–12。为此,研究人员实施了一系列相关策略来解决以上不足,如形貌晶格调控10,13,14,多级复合结构15–18和多金属合金化19–22等等。
金属有机框架(MOFs)材料具有大比表面积,可调谐的孔径和孔隙率,以及高度分散的不饱和金属中心等优点,其本身可以作为电催化剂,也可以作为前驱体进一步制备性能卓越的催化剂,因此具有广阔的应用前景23–25。当前,一系列的铜基26–29、锌基30、铁基31、钴基32,33、锆基33,34MOF已经被广泛用于二氧化碳电还原,但仍存在电流密度小、法拉第效率不高以及催化稳定性差等缺点。铜基MOF及其衍生物有着生成碳氢化合物,羧酸和醇类的特殊能力,是最具前景的CO2还原电催化剂。
通过在惰性气氛下热分解,MOF中的金属离子能够被转化为金属团簇,金属氧化物甚至是金属单原子,同时有机配体被碳化成多孔碳材料,能够有效吸附CO2分子并提供大量的催化活性位点35。例如,通过调控退火温度可以将HKUST-1转化为铜纳米团簇从而提高多碳产物的法拉第效率27,36,37。此外,将一些杂原子如B、N、P和S等掺入碳材料被证明能有效改变催化剂的电子状态配位结构。这些杂原子与碳原子结合可以形成单原子分散的M-X-C (M为中心金属离子,X为掺入的杂原子)活性位点来提升二氧化碳电还原的催化活性38。通常N作为主要的掺杂原子,B、P、S作为辅助掺杂原子,多原子协同能有效调控中间体的吸附和转化行为39。Huan等40设计制备了以FeN4为活性中心的Fe-N-C材料,在低电位下获得了高于90%的FECO。Ju等41研究了一系列的M-N-C (M =Mn、Fe、Co、Ni)材料催化二氧化碳还原时的反应活性、CO的转换效率及法拉第效率,证明Fe和Ni是这一系列中最为活泼的金属,其中Ni-N-C催化剂可以与Au和Ag催化剂相媲美。
对于Cu-N-C活性中心,尤其是由铜基MOF衍生得到的氮掺杂材料用于电化学还原二氧化碳的报道主要以外加氮芳香杂环修饰的HKUST-1及其衍生物为主42,存在副反应等问题,少有直接采用含氮有机物作为配体原位引入氮源。为此,我们采用2-氨基对苯二甲酸(BDC-NH2)为有机配体合成了一类新含氮MOF (Cu-NBDC)并以此衍生得到锚定在氮掺杂多孔碳上的Cu2O/Cu@NC电催化剂,通过不同手段对其进行了形貌和结构表征,测试其电还原二氧化碳的性能,研究了氮掺杂在ECRCO2中的影响。
2 实验部分
2.1 实验试剂和材料
泡沫铜(1.0 mm × 200 mm × 300 mm)购自广东烛光新能源科技有限公司。2-氨基对苯二甲酸(BDC-NH2,98%)与对苯二甲酸(BDC,98%)均购自安耐吉化学。氢氧化钾(KOH,AR),无水甲醇(CH3OH,AR)与N,N-二甲基甲酰胺(DMF,AR,≥99.5%)购自国药集团化学试剂有限公司。所有试剂均为分析纯,能直接使用不需要进一步纯化。超纯水由Sartorius arium mini reinstwasser-system制得。
2.2 电催化剂的制备
2.2.1 制备基于泡沫铜的氢氧化铜纳米线阵列Cu(OH)2 NA/CF
所有泡沫铜最先浸在丙酮中来除去表面的油污,然后用浓盐酸除去铜氧化层,最后用二次水冲洗,保存在惰性气氛下。Cu(OH)2NA/CF由在3mol·L-1氢氧化钾电解液中,15 mA·cm–2的电流密度下电化学阳极氧化方法氧化预处理的泡沫铜15 min制得,然后用二次水冲洗,最后在氮气气氛下干燥备用。
2.2.2 合成Cu-NBDC与Cu-BDC
1.656 mmol的2-氨基对苯二甲酸先溶解在10 mL的DMF中得到溶液。随后立刻加入2 mL的水和备用的一片Cu(OH)2NA/CF,在45 °C下溶剂热反应72 h。最终得到的Cu-NBDC粉末用DMF和无水甲醇离心洗涤干燥备用。Cu-BDC由不带氨基的对苯二甲酸替代了2-氨基对苯二甲酸为有机配体,实验方法与条件同上。
2.2.3 制备Cu2O/Cu@NC与Cu2O/Cu@C
多孔的Cu2O/Cu@NC 通过将之前得到Cu-NBDC MOF 前体在不同温度下以5 °C·min-1在Ar氛围(流量为100 mL·min-1)中退火一个小时得到,记作(Cu2O/Cu@NC)。Cu2O/Cu@C的制备条件同上,以此作为对照样品。
2.3 电催化剂的表征
2.3.1 材料表征
晶体结构由粉末衍射XRD (PXRD,Bruker D8 Advance , German)带 有 CuKαradiation (λ=0.154056 nm)表征。表面形貌和微观结构的表征采用配备能量色散X射线分析仪(EDX)的场发射扫描电子显微镜(SEM,Hitachi Model SU-8010,Japan)和场发射透射电子显微镜(TEM,FEI TECNAI G2 F20 200KV,USA)。样品表面元素的化学态的分析采用带有单色 AlKα(1486.6 eV)X射线光源的X射线光电子光谱仪(XPS,Thermo Fisher,USA,Escalab 250Xi)。拉曼光谱分析采用(Raman,Horiba HR-Evolution,USA)633 nm的激光激发。热重分析采用(SDT 2960,Japan),在氩气氛围下以10 °C·min-1的升温速率测试。
2.3.2 电化学表征
电化学还原二氧化碳反应(ECR-CO2)在H型电解池中进行。H型电解池分为两个部分,阴极区与阳极区用Nafion 117质子交换膜分隔。铂网(1 cm × 1 cm)和Ag/AgCl电极(饱和KCl)分别作为对电极和参比电极。参比电极距离工作电极0.5 cm。两个部分都装有30 mL的0.1 mol·L-1KHCO3溶液作为电解液。所有的工作电势均参考可逆氢电极(RHE):

通入饱和二氧化碳的电解液的pH值为6.8。所有电化学测试均采用电化学工作站(CHI760E,China)进行。取4.0 mg的催化剂材料加入800 μL乙醇和200 μL水及50 μL的5%的Nafion的混合溶液,密闭后超声分散制得催化剂浆料。取催化剂浆料20 μL均匀涂覆在在玻碳电极上,烘干后进行测试。
在测试前,阴极区的电解液通入氮气30 min来排尽空气。然后再通入高纯度的二氧化碳(99.999%)30 min。首先采用循环伏安法(CV)对催化剂进行活化,应用电势范围为0到-1.4 Vvs.RHE,扫速为100 mV·s-1。电化学比表面积(ECSA)在-0.05 – -0.1 Vvs.Ag/AgCl范围内,以10、25、50、100及200 mV·s-1的扫速测试得到,其计算公式为:
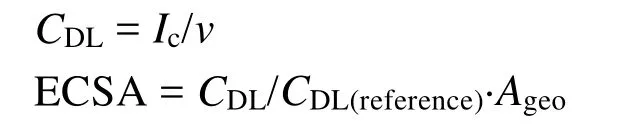
其中,CDL为催化剂的电化学双层电容,Ic为-0.075 Vvs.Ag/AgCl时同一扫速充电电流的平均值,v为扫速,CDL(reference)为空白玻碳电极的电化学双电容,Ageo为玻碳电极的几何面积(0.197 cm-2)。
气相色谱仪与H型电解池阴极区的封闭空间相连,测试中将含有剩余二氧化碳和气相产物的电解池流出相以高纯度氩气(99.999%)为载气在线注入气相色谱仪(GC,Agilent Technologies 7890B,USA)。液相产物则经核磁共振氢谱鉴定(1H NMR,Agilent Technologies DD2-600,USA)。产物的法拉第效率计算公式如下:

Z为由二氧化碳和水来生成一个分子要转移的电子数(如甲酸需要2个电子,乙醇需要12个,正丙醇需要18个等),n为产物的摩尔数,c是液相产物的浓度,可由产物的核磁标线(图S1与表S1,Supporting Information (SI))得到,V是电解液的体积,F为法拉第常数,I和t分别为电流和时间。
3 结果与讨论
制备Cu2O/Cu@NC与Cu2O/Cu@C的步骤如图1a所示,详细细节见实验部分。首先通过电化学阳极氧化,可以在泡沫铜(CF)表面均匀生长平均长度为7 μm,直径为200 nm的高密度氢氧化铜纳米线阵列(Cu(OH)2NA/CF)(图S2,(SI)所示)随后,我们以Cu(OH)2NA/CF作为可缓慢释放Cu2+的铜源,选择了三组常用溶剂:20 mL DMF,产物记作Cu-NBDC-D;等体积DMF与H2O共20 mL,产物记作Cu-NBDC-D-H;等体积DMF与EtOH共20 mL,产物记作Cu-NBDC-D-E,探索了溶剂对MOF生长的影响(图S3与图S4 (SI))。Cu-NBDC-D与Cu-NBDCD-E两者的形貌与晶体结构均相同,而Cu-NBDCD-H则不同,证明了此MOF合成中EtOH仅仅作为普通溶剂,而水则参与了配位或者自组装反应。最终选用DMF与水双溶剂在弱酸环境下可以生成球花形状的Cu-NBDC (本文Cu-NBDC均指Cu-NBDC-D-H)金属有机框架材料,同时泡沫铜表面的纳米线完全消失。改变溶剂中水的量能够有效调节Cu-NBDC的形貌(图S5 (SI))。当DMF与H2O体积比为100 : 1时得到均匀分布的褶皱状形貌;进一步增加水的体积比至5 : 1时则得到球花形状的Cu-NBDC。作为对照,我们用BDC替代BDC-NH2合成出的铜基MOF均为不规则的纳米片形貌,记作Cu-BDC。随后将两种MOF材料在不同温度下进行退火处理,得到了含氮的Cu2O/Cu@NC与不含氮的Cu2O/Cu@C。

图1 (a)以原位生长在泡沫铜基底上的Cu(OH)2纳米线为铜源生长Cu-NBDC与Cu-BDC,并经退火后得到Cu2O/Cu@NC与Cu2O/Cu@C复合材料;(b)Cu-NBDC,Cu-BDC分别与其配体BDC-NH2,BDC的红外光谱图;(c)上图为Cu-NBDC,下图为Cu-BDC的SEM图;(d)Cu-NBDC与Cu-BDC的XRD谱Fig. 1 (a)Scheme for the fabrication of Cu2O/Cu@NC and Cu2O/Cu@C by annealing Cu-NBDC and Cu-BDC grown on Cu(OH)2 NA/CF; (b)FTIR spectra of BDC-NH2 ligand and Cu-NBDC, BDC and Cu-BDC;(c)SEM images of Cu-NBDC (top)and Cu-BDC (bottom); (d)XRD patterns of Cu-NBDC and Cu-BDC.
图1b为两种铜基MOF与各自配体的红外光谱图。上半部为Cu-NBDC与BDC-NH2的红外光谱,下半部为Cu-BDC与BDC的红外光谱。从BDC-NH2与BDC的红外光谱中,我们可以在3200–2500 cm-1范围内观察到宽而散的羧羟基吸收带。在Cu-NBDC红外光谱的同样位置也能观测到明显的羧羟基吸收带,这表明Cu-NBDC中的羧基未完全参与配位。相比之下,羧基已经完全参与配位的Cu-BDC则几乎没有明显的羧羟基吸收带。在3504和3392 cm-1处对应于BDC-NH2中的νN-H,3427cm-1对应Cu-NBDC的νN-H,显示伯胺双峰转变为了仲胺的单峰,表明BDC-NH2在配位过程中氨基官能团发生了改变,参与铜离子的配位。图1c上部为Cu-NBDC的SEM图,能清晰看出其形貌为球花状,下部则为不规则片状形貌的Cu-BDC。图1d所示为两种MOF的晶体结构。两种MOF的XRD衍射峰均清晰且尖锐,意味着合成出的MOF晶型较好,且Cu-BDC衍射峰位与先前报道的Cu-BDC基本一致43。
Cu-NBDC与Cu-BDC的热重分析(TGA)见图S6 (SI)。从图中可以看出,Cu-NBDC在300 °C开始分解,350 °C分解结束,随后为碳氮氧的损失,且随着温度升高而增加。为此我们选择400、600和800 °C三个温度退火处理Cu-NBDC,分别记作Cu2O/Cu@NC-400、Cu2O/Cu@NC-600和Cu2O/Cu@NC-800。为了分析氮的掺入对ECR-CO2的影响,我们同样将Cu-BDC在这三个温度下进行退火。Cu-BDC的热重曲线可以分为三种明显的质量损失:溶剂的挥发,Cu-BDC中残留的水的脱去以及Cu-BDC的分解。图2a,b,c与d,e,f分别为400,600和800 °C三个退火温度处理的Cu2O/Cu@NC的SEM与TEM图;图2g,h,i与j,k,l分别为400,600和800 °C三个退火温度处理的Cu2O/Cu@C的SEM与TEM图(从左往右退火温度逐渐增加)。Cu-NBDC的球花状形貌在不同温度退火后依然能够保持的很好,而Cu-BDC的形貌则受到了破坏。退火后的样品表面变得粗糙,有大量熟化颗粒出现在其上(图S7与S8 (SI))。Cu2O/Cu@NC和Cu2O/Cu@C的TEM图像均显示了不同温度下形成了包覆Cu2O/Cu纳米颗粒的纳米结构。随后我们用XRD与Raman,BET等手段对Cu2O/Cu@NC与Cu2O/Cu@C进行了分析表征。图2m为这两种催化剂样品在不同温度下的XRD图,衍射峰在2θ=43.2°,50.3°和74.1°对应铜(JCPDS card No. 04-0836)的(111),(200)和(220)晶面。此外Cu2O/Cu@NC在36.3°,42.3°和61.2°对应氧化亚铜的(111),(200)和(220)晶面(JCPDS card No. 05-0667),Cu2O/Cu@C存在氧化亚铜的(111)晶面,表明两种催化剂同时存在氧化亚铜和铜。此外,可以明显看出随着温度的升高,Cu衍射峰增强,样品金属化程度增大。在所有Cu2O/Cu@NC样品中,拉曼光谱都检测到在280–620 cm-1之间的CuxN信号,并且CuxN的强度随着温度的升高而降低,这是由于在高温下N的流失(图2n)。与此同时,石墨碳在位于1336、1588 cm-1处有两条宽且重叠的D、G谱带,Cu2O/Cu@NC的D-G峰的分离随温度升高而增大,表明温度越高,碳化程度越高。而Cu2O/Cu@C-400则没有明显的D-G峰,这是因为Cu-BDC在400 °C时刚刚分解,此时碳层还没有完全形成,这与图S6中的TGA结果相一致。我们通过BET方法和N2吸脱附曲线分析了这两种催化剂在不同温度退火后的比表面积,Cu2O/Cu@NC的比表面积随退火温度先升高然后下降,由400 °C的230.24 m2·g-1增加到600 °C的278.49 m2·g-1,再降至800 °C的225.23 m2·g-1;而Cu2O/Cu@C的则随着退火温度升高而下降(图S9,表S2 (SI)所示),这可能是因为Cu2O/Cu@C在400 °C刚开始分解,MOF孔道还没有受到严重破坏,而随着温度升高孔道也逐渐被破坏坍塌。从N2吸脱附曲线来看,Cu2O/Cu@NC在较低相对压力(P/P0< 0.1)时吸附量迅速上升,在中高相对压力区域(0.4

图2 (a,b,c与d,e,f)分别为经不同温度退火的Cu2O/Cu@NC的SEM与TEM图;(g,h,i与j,k,l)分别为不同温度退火的Cu2O/Cu@C的SEM与TEM图(从左往右温度逐渐增加);(m,n)分别为经不同温度退火的Cu2O/Cu@NC与Cu2O/Cu@C的XRD衍射谱和拉曼光谱图Fig. 2 (a, b, c, and d, e, f)SEM and TEM images of Cu2O/Cu@NC; (g, h, i, and j, k, l)SEM and TEM images of Cu2O/Cu@C annealed at different temperature; (m, n)XRD patterns and Raman spectra of Cu2O/Cu@NC-400,Cu2O/Cu@NC-600, Cu2O/Cu@NC-800, Cu2O/Cu@C-400, Cu2O/Cu@C-600 and Cu2O/Cu@C-800.
我们用H型三电极体系,在CO2饱和的0.1 mol·L-1KHCO3电解液中评估了不同温度下制得的催化剂Cu2O/Cu@NC与Cu2O/Cu@C的电化学还原二氧化碳反应的活性。我们首先在-0.05 – -0.1 Vvs.Ag/AgCl范围内,以10、25、50、100及200 mV·s-1的扫速评估了不同样品的电化学比表面积(ECSA)(图S10与表S3 (SI)),结果显示Cu2O/Cu@NC的ECSA均大于Cu2O/Cu@C,其中Cu2O/Cu@NC-400的ECSA最大,为1.23 cm-2。随后,我们对不同样品进行循环伏安曲线(CV)测试,如图3a所示,Cu2O/Cu@NC的电流密度在相应的退火温度下均要高于Cu2O/Cu@C,其中Cu2O/Cu@NC-400在所有样品中的电流密度最大,说明其具有最佳的电催化效率。

图3 Cu2O/Cu@NC-400,Cu2O/Cu@NC-600和Cu2O/Cu@NC-800,Cu2O/Cu@C-400,Cu2O/Cu@C-600和Cu2O/Cu@C-800在含饱和二氧化碳的0.1 mol·L-1 KHCO3电解液中的电化学还原二氧化碳反应Fig. 3 Performance of electrochemical CO2 reduction by Cu2O/Cu@NC-400, Cu2O/Cu@NC-600 and Cu2O/Cu@NC-800,Cu2O/Cu@C-400, Cu2O/Cu@C-600 and Cu2O/Cu@C-800 in CO2-saturated 0.1 mol·L-1 KHCO3 electrolyte.
为了进一步评估两种催化剂研究的CO2电还原催化性能,讨论N掺杂的影响。我们在0.8 – -1.6 Vvs.RHE的电势范围内详细讨论了不同样品的CO2还原产物的种类以及选择性。其中,Cu2O/Cu@NC的气相产物为C2H4、CH4、CO,Cu2O/Cu@C的为CH4、CO,均伴随着氢气的析出。两种催化剂的液相产物均主要为甲酸盐(formate)。从图3b,c以及3d及表1来看,两种类型催化剂的甲烷法拉第效率均随着过电势的升高而增加,而产物甲酸盐随着过电势的增加先升高后降低,在-1.2 Vvs.RHE时法拉第效率达到最大。图3e为甲酸盐的分电流密度曲线图,我们可以看出甲酸盐的分电流密度随着电势的增加而增加。此外,不同温度下的Cu2O/Cu@NC均在-1.4 Vvs.RHE时达到最大乙烯法拉第效率(图3b),且C2H4与CH4的法拉第效率随着Cu2O/Cu@NC退火温度的升高而下降,而甲酸盐的法拉第效率逐渐增加。其中,Cu2O/Cu@NC-400的乙烯和甲烷的法拉第效率最大,分别为FEC2H2= 20.4%和FECH4= 23.9%。Cu2O/Cu@C则无明显乙烯和甲烷的生成。因此我们认为氮的掺杂显著影响了其CO2催化路径,向乙烯和甲烷产物转变。由图3f的氢气法拉第效率(FEH2)图可以看出,在同样电位下Cu2O/Cu@NC的FEH2明显小于Cu2O/Cu@C,Cu2O/Cu@C-800的最大FEH2已经高达71.1%。此外,Cu2O/Cu@NC-400的CO2催化效率在-1.4 – -1.6 Vvs.RHE范围内高于86%,而Cu2O/Cu@C的二氧化碳还原效率最高不足50%。由此我们可以看出掺入氮的催化剂Cu2O/Cu@NC对氢气的抑制效果非常明显。
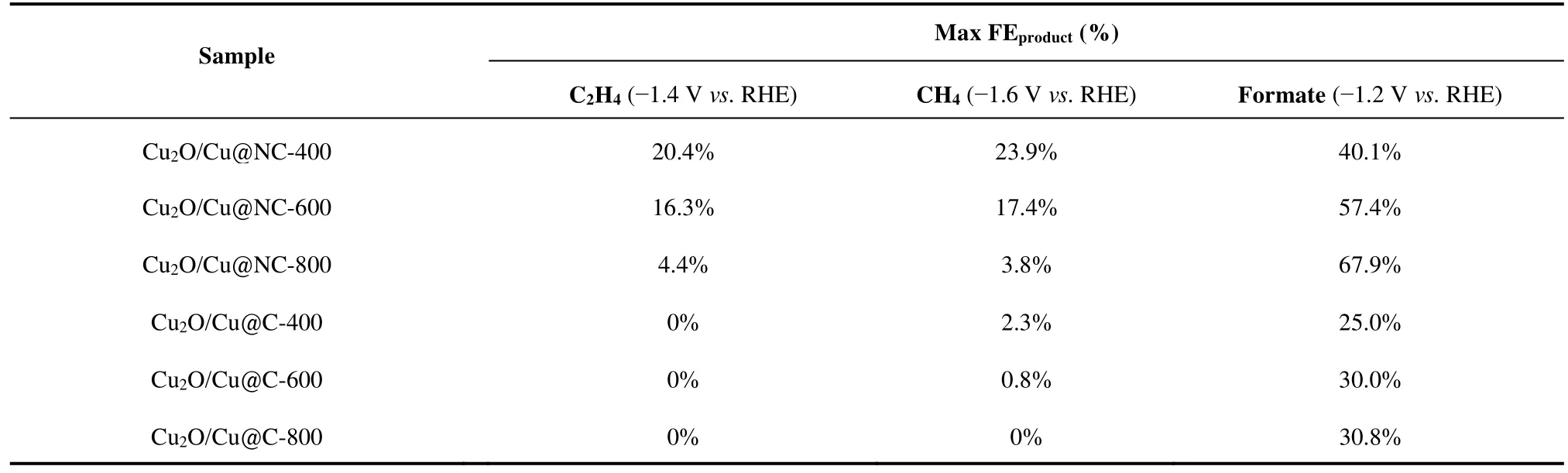
表1 不同温度退火的Cu2O/Cu@NC和Cu2O/Cu@C的二氧化碳电还原性能Table 1 Performance of CO2 electrochemical reduction for Cu2O/Cu@NC and Cu2O/Cu@C annealed at different temperatures.
为了分析造成CO2电化学活性如此显著差异的原因以及氮掺杂的作用,我们采用了XPS来定量分析不同退火温度的Cu2O/Cu@NC与Cu2O/Cu@C样品的元素组成和化学态。由图4a中的Cu2O/Cu@NC-400的XPS全谱可知,Cu2O/Cu@NC样品含有铜、碳、氮和氧四种元素。图4b为Cu2O/Cu@NC的Cu 2p3/2XPS光谱(从上往下退火温度逐渐增加),图S11 (SI)为Cu2O/Cu@C的Cu 2p3/2XPS光谱,在932.2 eV处可以分解为两个对应于Cu(0)和Cu(I)的子峰46,可以明显看到,复合的纳米粒子主要为Cu2O和金属Cu,且随着退火温度的升高,总Cu含量也在增加(表S4,S5 (SI)),Cu(I)的百分比含量在逐渐下降而Cu(0)的含量逐渐增加(图S12、表S6 (SI)),这与XRD的结果相一致(图2m)。这表明随着退火温度的升高,碳对铜离子的还原性越强,铜离子被还原为结晶度更高的金属铜单质。由表S4与S5可知,Cu2O/Cu@NC与Cu2O/Cu@C在相同温度处理下的总铜含量近似相同,但结合表1中二氧化碳电还原反应产物的最大法拉第效率来看,铜含量对CO2RR最终产物的选择性影响不大。图4c显示了不同温度处理的Cu2O/Cu@NC的C 1s高分辨XPS光谱,C 1s峰可分为C=C/C-C (284.4 eV),C-N (285.2 eV),COH (286.2 eV)和C=O (288.4 eV)。C-N键的存在意味着BDC-NH2中的氮原子被掺入到碳基体中。随着退火温度升高,石墨碳的比例相应增加,这与拉曼的测试结果吻合。N 1s的高分辨光谱和不同退火温度样品的N含量见图4d,e。所有N 1s光谱都能够分为至少六个峰:pyridinic-N (398.2 eV),Cu-N (399.0 eV),pyrrolic-N (399.8 eV),graphitic-N(400.3 eV),quaternary-N (401.2 eV)和oxidized-N(402.5 eV)。N可以合并到Cu2O的氧晶格中47,因此Cu-N键的存在证明N已掺入到Cu2O晶格中46,48。随着退火温度的升高,总N含量由Cu2O/Cu@NC-400的9.09%升到Cu2O/Cu@NC-600的11.42%,又降至Cu2O/Cu@NC-800的5.57% (表S4)。高温退火的样品中Cu-N和pyrrolic-N的含量降低而oxidized-N的含量显著增加(表S7 (SI))。这可能是因为稳定的N保留在碳晶格中,而不稳定的N原子在高温下容易被氧化或损失,导致氧化态N含量增加49–51。

图4 (a)Cu2O/Cu@NC-400的XPS全谱;(b,c)Cu2O/Cu@NC的高分辨XPS Cu 2p3/2和C 1s谱(从上往下退火温度逐渐增加);(d)N物种及含量分析;(e)Cu2O/Cu@NC-400,Cu2O/Cu@NC-600和Cu2O/Cu@NC-800的XPS N 1s高分辨光谱Fig. 4 (a)XPS survey spectrum of Cu2O/Cu@NC-400; High-resolution XPS spectra of (b)Cu 2p3/2 and(c)C 1s for Cu2O/Cu@NC annealed at different temperature; (d)Summary of various N species and contents;(e)High-resolution N 1s spectra of Cu2O/Cu@NC-400, Cu2O/Cu@NC-600 and Cu2O/Cu@NC-800.
在二氧化碳电还原过程中有两个重要的中间产物:*OCHO和*COOH。*COOH反应途径可以进一步被还原为*CO,能够在质子-电子化作用下生成*CH2,这是加氢生成甲烷或者发生C-C偶联反应二聚成乙烯的重要中间体,而*OCHO途径则能生成甲酸盐45,52。我们分析了乙烯和甲烷的最大法拉第效率与不同种类氮的关系,从图S13 (SI)可以看出,乙烯和甲烷的最大法拉第效率与Cu-N及pyrrolic-N含量呈正相关关系,而其他种类的N则没有这种趋势。然而,Sharma等53通过理论计算(DFT)证明了pyrrolic-N对电化学还原二氧化碳的影响很小或者没有影响。由此,我们认为Cu-N含量的增加提高了CO2转化成乙烯和甲烷的选择性。随着退火温度的升高,Cu-N含量在减少,而甲酸盐的最大法拉第效率在增加。结合以上发现,我们推测Cu-N的含量能够影响二氧化碳电还原的反应路径(图5),当Cu-N含量增加时,二氧化碳还原反应选择了Path I,即先生成*COOH,进一步还原为*CO,随后在质子-电子化作用下生成*CH2,从而进一步生成乙烯和甲烷;当Cu-N含量减少时则选择了先生成*OCHO再生成HCOOH (Path II)。Cu-N的存在稳定了二氧化碳还原反应中*CH2中间体的吸附,抑制了*H生成氢气。与不含氮的Cu2O/Cu@C相比,Cu2O/Cu@NC催化剂提升了二氧化碳还原反应的整体催化性能。为了更深入地了解N的掺杂是如何通过调整Cu2O的电子结构来改变还原产物的选择性,需要进一步开展原位表征实验和第一性原理计算。

图5 二氧化碳电还原反应可行的路径Fig. 5 The possible paths for electrochemical CO2 reduction.
4 结论
综上所述,通过对Cu-NBDC在不同温度下退火,我们成功地得到将Cu2O/Cu纳米颗粒锚定在N掺杂的多孔碳上的催化剂(Cu2O/Cu@NC)。与不含氮的Cu2O/Cu@C相比,氮的掺入显著提升了二氧化碳还原反应的整体催化性能。碳基体中Cu-N的存在稳定了二氧化碳还原反应中*CH2中间体的吸附,从而有效抑制氢气的析出,促进了催化剂表面乙烯和甲烷的生成,并且Cu-N含量越高,Cu2O/Cu@NC对乙烯和甲烷的选择性越高。在400 °C时分别获得了20.4%的乙烯法拉第效率及23.9%的甲烷法拉第效率。随着退火温度的升高,Cu-N含量降低,二氧化碳还原反应路径变为朝着生成甲酸盐的方向进行,所制备的催化剂(Cu2O/Cu@NC-800)对电化学还原二氧化碳成甲酸盐具有良好的选择性,甲酸盐的最高法拉第效率达到67.9%。我们的研究证明了氮的掺入能显著提高铜基催化剂的ECR-CO2催化性能,有效改变CO2催化路径,这为今后拓展制备MOF衍生的高效铜基催化剂提供可参考的策略。
Supporting Information:available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
猜你喜欢
核化学与放射化学(2022年2期)2022-04-28
军民两用技术与产品(2021年10期)2021-03-16
水上消防(2020年1期)2020-07-24
小学生必读(高年级版)(2019年3期)2019-06-21
疯狂英语·新读写(2018年3期)2018-11-29
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
作文周刊·七年级版(2016年47期)2017-05-31
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
故事会(2015年12期)2015-05-14

- 物理化学学报的其它文章
- Microscopic Mechanism on Giant Photoeffect in Proton Transport Through Graphene Membranes
- 二维材料范德华间隙的利用
- 全固态电池界面的研究进展
- Synthesis, Characterization, and Crystal Structure of Lithium Pyrrolide
- Gas-Phase Mechanism Study of Methane Nonoxidative Conversion by ReaxFF Method
- Single-Atom Cobalt Coordinated to Oxygen Sites on Graphene for Stable Lithium Metal Anodes