
Synthesis, Characterization, and Crystal Structure of Lithium Pyrrolide
2021-11-22 07:01ZijunJingKhaiChenTanTengHeYangYuQijunPeiJintaoWangHuiWuPingChen
物理化学学报 2021年11期
Zijun Jing , Khai Chen Tan , Teng He , Yang Yu , Qijun Pei , Jintao Wang , Hui Wu ,Ping Chen
1 Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning Province, China.
2 University of Chinese Academy of Sciences, Beijing 100049, China.
3 NIST Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD 20899-6102, USA.
Abstract:Development of clean energy is an urgent requirement because of the depletion of fossil energy sources and increasingly severe environmental pollution.However, the lack of safe and efficient hydrogen storage materials is one of the bottlenecks in the implementation of hydrogen energy. Liquid organic hydrogen carriers (LOHCs)have been recognized as potential materials for the storage and transportation of hydrogen owing to their high gravimetric and volumetric hydrogen densities, reversible hydrogen absorption and desorption ability, and ease of widespread implementation with minimal modification on the existing fueling infrastructure. While some LOHCs such as cycloalkanes and N-heterocycles have been developed for hydrogen storage, they require a high hydrogen release temperature due to the large enthalpy change of dehydrogenation. In our previous work, a metallation strategy was proposed to improve the thermodynamic properties of liquid organic hydrogen carriers for hydrogen storage, and a series of metalorganic hydrides were synthesized and investigated. Among them, sodium phenoxide-cyclohexanolate pair, lithium carbazolide-perhydrocarbazolide, and sodium anilinide-cyclohexylamide pair showed promising dehydrogenation thermodynamics and improved hydrogen storage properties. Sodium pyrrolide and sodium imidazolide were also synthesized. However, pyrrolides were not well characterized, and the structure of lithium pyrrolide was not resolved. In the present study, we synthesized sodium and lithium pyrrolides by ball milling and wet chemical methods. One equivalent of hydrogen could be released from the reaction of pyrrole and metal hydrides, indicating the replacement of H by metal.The formation of pyrrolides was confirmed by nuclear magnetic resonance (NMR), X-ray diffraction (XRD)and ultravioletvisible spectroscopy analyses. The 1H signals attributed to C-H in the NMR spectra of the alkali metal pyrrolides shifted upfield due to the replacement of the H of N-H with a stronger electron-donating species (Li or Na), resulting in a greater shielding environment upon metallation. The absorption peaks of lithium and sodium pyrrolides showed red shifts, and the intensities became obviously stronger in the UV-Vis spectra, suggesting an enhancement of the conjugation effect, in accordance with theoretical calculations. The structure of lithium pyrrolide was determined by the combined direct space method and first-principles calculations on XRD data and Rietveld refinement. This molecule crystallizes in the monoclinic P21/c (14)space group, with lattice parameters of a = 4.4364(7)Å, b = 11.969(2)Å, c = 8.192(2)Å, β = 108.789(8)°, and V = 411.8(2)Å3 (1 Å = 0.1 nm). Each Li+ cation is surrounded by three pyrrolides via cation-N σ bonding with two pyrrolides and a cation–π interaction with the third pyrrolide, where the Li+ is on the top of the π face. Our experimental findings are different from the theoretical prediction in the literature.
Key Words:Lithium pyrrolide;Crystal structure;Metal replacement;Liquid organic hydrogen carrier;Metallo-N-heterocycle;Hydrogen storage
1 Introduction
The development of clean energy is urgently necessary due to the exhausted fossil energy sources and increasingly severe environmental pollution. Hydrogen as an energy carrier has been widely studied because of its high energy density and cleanliness1,2. However, lack of safe and efficient hydrogen storage materials is one of the bottlenecks for the implementation of hydrogen energy. In the past decades,tremendous efforts on hydrogen storage have been devoted to inorganic hydrides, for instances, metal hydrides3, chemical hydrides2,4,5, and complex hydrides6–9. Nevertheless, they encountered low hydrogen capacity, poor reversibility or inferior thermodynamics. Recently, organic hydrides with the advantages of low cost, safe, and moderate hydrogen capacity were drawing much attention, which may have potential to meet the U.S. Department of Energy’s (DoE)targets10–13. The cycloalkanes were proposed for hydrogen storage initially,including benzene, methylbenzene, naphthalene,etc. However,the enthalpies changed of dehydrogenation (ΔHd)of cycloalkanes are high, resulting in relatively high dehydrogenation temperatures. To optimize the thermodynamics, it was confirmed experimentally and theoretically by Crabtree14and Pez15et al. that the incorporation of heteroatoms into organic hydrides reduced the ΔHdand dehydrogenation temperatures to a certain extent. It was reported that cyclic organic hydrides with electron donating groups outside the carbon-rings showed lower dehydrogenation enthalpy changes16. Very recently, we proposed a metallation strategy,i.e., replacing the active hydrogen atom in organic hydrides by alkali or alkaline earth metals, and a series of metalorganic hydrides with superior thermodynamic properties were successfully developed17–19. For instance, the ΔHdof N-heterocycles could be reduced efficiently by forming metallo-N-heterocycles, where the optimized thermodynamics is due to the electron donation from the metal to organic ring. Among them,alkali pyrrolides as examples of metallo-N-heterocycles were synthesized by ball milling pristine pyrrole and corresponding alkali hydrides. However, the pyrrolides were not well characterized and the crystal structure of lithium pyrrolide was not resolved until now. Although its structure and cation–πinteraction were predicted theoretically by Rodgers and Elguero20,21, further validation and confirmation from experiment are highly needed. In addition, considering that pyrrolide is often used as an intermediate in organic synthesis22because of its special properties of pyrrole ring23, the synthetic routes to pyrrolides should be exploited and broadened.Therefore, in the present study, two methods were employed to synthesize lithium and sodium pyrrolides which were further characterized in detail. On account of the detailed characterization, the crystal structure of lithium pyrrolide was successfully resolved.
2 Experimental
Pyrrole (98%, Alfa Aesar), lithium hydride (97%, Alfa Aesar)and sodium hydride (90%, Aldrich)were commercially purchased and used without further purification. Diethyl ether(AR, Kermel)was used after removal of water with molecular sieve.
Ball milling method and wet chemical method were used to synthesize lithium and sodium pyrrolides. All the experiments were conducted in a glove box filled with purified argon to avoid contamination by air.
Ball milling method: pyrrole and lithium hydride/sodium hydride were mixed in 1 : 1 molar ratio in a 180 mL stainless steel vessel. The mixture was pre-milled in a Retsch PM 400 planetary ball mill at a rate of 60 r·min-1for 0.5 h. For the synthesis of lithium pyrrolide, the sample was further milled at a rate of 150 r·min-1for 4 h at 50 °C. While for sodium pyrrolide,it was milled at a rate of 200 r·min-1for 4 h at room temperature.Around 1 equivalent of hydrogen was released from both reactions, which were determined by pressure change in the vessel and mass spectroscopy (MS).
Wet chemical method: pyrrole and lithium hydride with molar ratio of 1 : 1 were added into a 60 mL autoclave, then 20 mL diethyl ether was added, followed by the mixture was stirred at a rate of 500 r·min-1at 65 °C. The reaction was monitored by a pressure gauge. After 48 h, approximately 1 equivalent of hydrogen was released and determined by MS. Finally, the purified product was obtained after the removal of diethyl ether by distillation under reduced pressure.
The synthesized lithium pyrrolide and sodium pyrrolide were characterized by X-ray diffraction (XRD), ultraviolet-visible(UV-Vis)spectrophotometry, liquid-state nuclear magnetic resonance (NMR)and magic angle spinning NMR (MAS NMR).XRD patterns were collected on a PANalytical X’Pert diffractometer equipped with CuKαradiation at 40 kV and 40 mA. UV-Vis spectroscopy was collected on a Jasco V-750 spectrophotometer. Liquid state and MAS NMR spectroscopies were obtained on a Bruker AVANCE 500 MHz NMR spectrometer (11.7 T)at room temperature and DMSO-d6 was used as a deuterated reagent.
3 Results and discussion
3.1 Syntheses of lithium and sodium pyrrolides
Lithium pyrrolide and sodium pyrrolide were synthesizedviaball milling pyrrole with lithium hydride and sodium hydride,respectively. Around 1 equivalent of hydrogen was released from both reactions (experimental part). Only hydrogen was detected in the gaseous products by MS after the reaction, indicating the formation of lithium pyrrolide and sodium pyrrolide following reactions (1)and (2). For the synthesis of lithium pyrrolide, wet chemical method was employed, where 1 equivalent of hydrogen was evolved as shown in Fig. 1. After distillation under reduced pressure, a white powder was obtained, which is similar to that from ball milling method.


Fig. 1 Hydrogen evolution from the reaction of lithium hydride and pyrrole in diethyl ether solvent. Inset figure is MS signals of gaseous products. Except H2, other signals come from the solvent diethyl ether.
3.2 Characterizations
NMR was employed to investigate the chemical state of synthesized pyrrolides in DMSO-d6. As shown in Fig. 2a, H―N signal at 10.75 ppm can be clearly observed in pristine pyrrole.By comparison, the1H signals of N―H in alkali metal substituted pyrrolides disappeared, strongly indicating that the alkali metals have replaced the active hydrogen on N.Furthermore, the1H signals of C―H in alkali metal pyrrolides shifted upfield which reveal the increased of electron density of C-rings. The upfield shift of1H signal can be attributed to the replacement of H of N―H with a more electron donating element (Li or Na), thus creating a more shielding environment upon metallation. Meanwhile, the shifted resonances of13C also suggest the impact of alkali metals replacement (Fig. 2b).However, the C atoms which close to and far from N atom showed downfield and upfield shift, respectively, agreeing well with our previous theoretical simulation18.
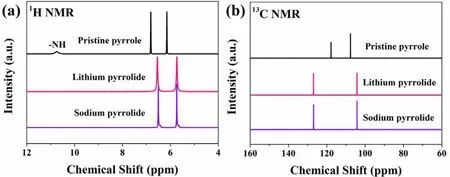
Fig. 2 1H and 13C NMR spectra of synthesized lithium pyrrolide, sodium pyrrolide and pristine pyrrole.
Additionally, liquid state7Li and23Na NMR spectra of synthesized lithium and sodium pyrrolides in DMSO-d6exhibited obvious7Li and23Na signals at 0.27 and 3.84 ppm,respectively (Fig. 3a), indicating the formation of soluble lithium and sodium salts.7Li MAS NMR on lithium pyrrolide exhibited an upfield shift in comparison to pristine LiH (Fig. 3b).Therefore, NMR results suggested the replacement of H on N with alkali metals could affect the chemical environment of pyrrole ring.

Fig. 3 (a)Liquid state 7Li and 23Na NMR spectra of synthesized pyrrolides. (b)7Li MAS NMR spectrum of synthesized lithium pyrrolide in comparison with pristine LiH.
The XRD patterns of synthesized lithium pyrrolide and sodium pyrrolide were shown in Fig. 4. The result of sodium pyrrolide is in good agreement with the previous report18,showing the formation of targeted product. Lithium pyrrolides,both from ball milling and wet chemical method exhibited similar diffraction patterns, which were different from pristine LiH, suggesting the formation of new phases.

Fig. 4 XRD patterns of synthesized lithium pyrrolides and sodium pyrrolide as compared to pristine LiH.
As shown in UV-Vis spectra (Fig. 5), the absorption peak of pristine pyrrole was 256.0 nm. In comparison, the absorptions of lithium pyrrolide and sodium pyrrolide demonstrated red shifts and the intensities became obviously stronger,i.e., the wavelengths at 259.0 and 259.2 nm were attributed to lithium and sodium pyrrolides, respectively, suggesting the enhancement of conjugation effect. Moreover, it was found that the conjugation effect in sodium pyrrolide is slightly stronger than lithium pyrrolide, agreeing well with our theoretical calculation results that sodium could donate more electron to pyrrole ring18. Therefore, the formation of alkali pyrrolides was further demonstrated.

Fig. 5 UV-Vis spectra of synthesized lithium pyrrolide and sodium pyrrolide correlated to pyrrole in DMSO-d6.
3.3 Crystal structure
XRD was used to identify the crystal structure of lithium pyrrolide as shown in Fig. 6. The XRD pattern of lithium pyrrolide was indexed using a monoclinicP21/c(14)space group structure with lattice parameters of approximatelya= 4.431 Å,b= 11.968 Å,c= 8.193 Å (1 Å = 0.1 nm), andβ= 108.75°. The crystal structure was then partially solved using direct space methods under this space group. Due to the uncertain H and Li positions, first-principles molecular dynamics simulated annealing were then performed to confirm the C4H4N and Li configuration and coordination with the lowest energy. Rietveld structural refinement on the optimal structural candidate was performed using the GSAS package on the XRD data. The C4H4N were kept as rigid bodies with relaxed bond lengths and bond angles constrained due to inadequate number of observations. The rigid body and Li coordination together with lattice parameters were refined, yielding the agreement factors ofRwp= 0.0500,Rp= 0.0387 andχ2= 1.527 (RwpandRpare weighted profile and profileR-factors, respectively.χmeans the ratio of weighted profile to expected profileR-factors. TheR-factors andχ2functions are used to evaluate the quality of fit to a powder diffraction pattern). The Rietveld fit to the XRD pattern was shown in Fig. 6 and the refined lattice parameters of LiC4H4N were:a= 4.4364(7)Å,b= 11.969(2)Å,c= 8.192(2)Å,β= 108.789(8)° andV= 411.8(2)Å3.

Fig. 6 Experimental (circles), fitted (line), and difference (line below observed and calculated patterns)XRD profiles for lithium pyrrolide LiC4H4N at 298 K. Vertical bars indicate the calculated positions of Bragg peaks.
The crystal structure of lithium pyrrolide and the local coordination of the Li+cation were shown in Fig. 7. Each Li+cation is surrounded by three pyrrolides, interacting with two of them through cation―Nσbonding and the thirdviacation–πinteraction with the Li+on top ofπface of pyrrolide ring. The two neighboring strand of pyrrolide anions were interlaced together alternatively through both Li―Nσbonding and the cation–πinteraction, forming a braid of Li-pyrrolide 1D arrangement alongaaxis (Fig. 7a). The crystal structure is composed of such double-chain strings of pyrrolide rings. They interact with neighboring strings through the weak Van de Waals interaction, and spread alongbandcaxes, forming a 3D arrangement. The distances between Li and N inσbond are in the range of 2.0983 to 2.1639 Å (note: bond lengths are from the fully relaxed structure), which are shorter than that in the cation–πinteraction (Table 1). Most of the bonds including N―C, C=C, C―C and C―H are elongated upon metallation, which is in accordance with the theoretical results that the electron donation from alkali metals to pyrrole causes the bond elongation in pyrrole20,24.

Fig. 7 (a)Crystal structure of lithium pyrrolide and (b)coordination geometry of Li+ cation. Yellow sphere: lithium; blue sphere: nitrogen;red sphere: carbon; gray sphere: hydrogen.

Table 1 Interatomic distances (Å)comparison of lithium pyrrolide with pristine pyrrole and theoretical calculated lithium pyrrolide and pyrrole.
There have been theoretical studies on the coordination mode of Li+cation and pyrrolide20. According to the theoretical results, lithium pyrrolide may be more stable when the cation―N interaction is in aπconfiguration than in aσbonding because of the large orbital interaction between the lone pair of N with the C―C antibonding orbital in the cation–πconfiguration. As a result, the lithium pyrrolide would be more aromatic. However, from our experimental result, we found the presence of both cation―Nσbonding and cation–πinteraction in lithium pyrrolide, and the distance of Li+―N in the cation–πinteraction is much longer than that from theoretical simulation(Table 1). The reason may lie in that the theoretical result in the literature was obtained from the gas phase simulation and the only one molecule was considered for the Li―Nσbonding or cation–πinteraction. However, we resolved the crystal structure of lithium pyrrolide considering all the interactions in the 3D arrangement.
4 Conclusions and Outlooks
In summary, we synthesized sodium and lithium pyrrolides by ball milling method and wet chemical method, which were confirmed by NMR, XRD and UV-Vis. Lithium pyrrolide crystallizes in monoclinicP21/c(14)space group, with the lattice parameters ofa= 4.4364(7)Å,b= 11.969(2)Å,c= 8.192(2)Å,β= 108.789(8)° andV= 411.8(2)Å3. Each Li+cation is surrounded by three pyrrolides,viacation―Nσbonding with two pyrrolides and a cation–πinteraction with the third. Our experimental findings are different from the theoretical prediction in literatures. Further investigation is needed to broaden its applications, such as ionic conductivity, organic synthesis and so on.

- 物理化学学报的其它文章
- Microscopic Mechanism on Giant Photoeffect in Proton Transport Through Graphene Membranes
- 二维材料范德华间隙的利用
- 全固态电池界面的研究进展
- Gas-Phase Mechanism Study of Methane Nonoxidative Conversion by ReaxFF Method
- 含氮金属有机框架衍生的铜基催化剂电催化还原二氧化碳
- Single-Atom Cobalt Coordinated to Oxygen Sites on Graphene for Stable Lithium Metal Anodes