
金属有机骨架材料在二氧化碳加氢中的应用
2021-11-20 08:33:16南永永田海锋唐小华
燃料化学学报 2021年10期
周 程 ,南永永 ,查 飞,* ,田海锋 ,唐小华 ,常 玥
(1. 西北师范大学 化学化工学院,甘肃 兰州 730070;2. 靖远煤业集团刘化化工有限公司,甘肃 白银 730900)
近年来,随着工业的快速发展和过量化石燃料燃烧,使大气中的二氧化碳浓度逐年增加,导致了许多环境问题,包括温室效应、海洋酸化等,如何将CO2资源化利用引起了科研工作者的广泛关注[1]。CO2可作为碳源与可再生能源如太阳能、风能、生物质能等制得的氢气进行反应,生成高附加值的化学品,例如一氧化碳、甲醇、碳氢化合物、环状碳酸酯、恶唑烷酮等[2,3]。用于CO2化学转化的催化剂包括沸石[4]、无机盐[5]、有机配合物[6]、离子液体[7,8]和有机物框架材料[9]等。金属有机骨架材料(MOFs)由于其大的比表面积和孔体积、独特的孔道结构以及表面的酸碱性能,可用于气体吸附[10]、分离[11]、传感[12]、催化[13]等方面。MOFs 作为一种温和高效的催化剂,在二氧化碳的化学转化中取得较好的结果[14–17]。在CO2加氢系统中,研究最多MOFs 材料有Zn、Co 基ZIFs 系列(ZIF-8、ZIF-67)、Zr 基UiO 系列(UiO-66、UiO-67),MIL 系列(MIL-53、MIL-101)、Cu 基MOFs(Cu-BTC) 等。实际上,新型和有效的非均相催化剂用于二氧化碳催化加氢的研究引起了越来越多的兴趣,并且发表的许多评论总结了该领域传统催化材料的最新进展[15–21]。CO2加氢也是生产有价值的化学品的有效方法[22]。因此,本篇综述系统地总结了基于MOFs 作为催化剂促进CO2加氢转化的最新研究成果,分析了基于MOFs 的催化剂的合成方法和高催化活性的原因,讨论了MOFs 面临的一些问题和挑战,提出了提高基于MOFs 的材料催化CO2化学转化性能的可行策略。
1 MOFs 在CO2 加氢中的应用
1.1 CO2 加氢制一氧化碳
一氧化碳是合成气和各种煤气的主要成分,还用作合成甲醇[23,24]、乙酸[25]和光气[26]的重要C1 原料,并在冶金工业中用作还原剂[27]。通过逆水煤气变换(RWGS) 反应将CO2催化转化为CO,即CO2+ H2= CO + H2O,通常被认为是CO2加氢转化有前途的方法之一。目前,RWGS 反应的研究主要集中在贵金属催化剂(如Pt、Au 和Pd)的研究上,对非贵金属催化剂的研究报道很少。近几十年来,低温和高活性RWGS 催化剂的设计和探索引起了研究人员的极大关注。尽管在MOF 在催化领域已经取得了许多成就,但是使用MOF 将CO2加氢还原为CO 的研究才刚刚开始。Xu 等[28]基于Zr(IV)的MOF(UiO-67)支撑的金纳米颗粒催化剂(AuNPs@UiO-67)是MOF 在RWGS 反应中应用的第一个例子。由于AuNPs@UiO-67 复合催化剂在UiO-67 骨架表面上具有高度分散的Au NPs(金纳米颗粒),在固定床反应器中,反应条件为405 ℃ 、H2/CO2= 3,该催化剂的CO2转化率为30.5%,CO选择性为96.5%。此外,寿命实验表明该催化剂具有较高的热稳定性,并且在MOF 的表面上没有聚集Au NPs。众所周知,通过MOF 前体可以直接热解来制备MOF 衍生物,即金属(氧化物) / 碳纳米复合物,它往往具有独特的催化活性和稳定性。Zhang 等[29]在500 ℃的温度下以ZIF-8 为牺牲模板,和均苯三甲酸金属络合物(Cu-BTC)通过溶剂热合成法制备了CuZn-BTC CP 纳米棒,然后煅烧和还原获得多孔的分层Cu/Zn@C 杂化复合材料(图1),在H2/CO2= 3,t= 500 ℃,p= 0.1 MPa 的反应条件下,实现了100% 的CO 选择性和5.0%的CO2转化率,亚毫米尺寸的Cu/Zn@C 比Cu@C、Cu/SiO2,Cu-Zn-Al 催化剂表现出更高的CO2转化率和CO 选择性。他们发现将Cu/Zn 封装到MOF基质中可防止纳米颗粒烧结并产生协同效应,并且铜和锌的协同作用对水煤气的反向转换是有益的。他们为MOFs 转变为高性能催化剂提供了机会。
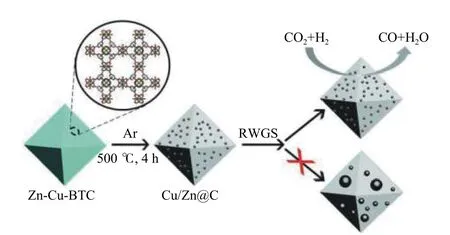
图1 ZIF-8 热解合成Cu/Zn@C 示意图[29]Figure 1Pyrolysis ofZIF-8t o produce hierarchical Cu/Zn@C[29]
另一个Zr 基的MOF UiO-66 [Zr6O4(OH)4(BDC)6]被用来制备单分散球形Pt/Au @Pd@UiO-66 配合物的模板,Zheng 等[30]将Au@Pd 双金属纳米颗粒包裹在UiO-66 的核中,然后将Pt 纳米颗粒涂覆在UiO-66 上,构建了新型复合Pt/Au@Pd@UiO-66(图2)。在固定床流动反应器中对所获得的催化剂在不同温度下进行了测试,结果表明,升高温度有利于CO2的转化,在400 ℃下,Pt/Au@Pd@UiO-66 的CO2转化率为35.3%。但是CO 的选择性随温度的升高而降低,这主要是由于RGWS 反应吸热和CH4含量的增加。将活性金属纳米颗粒组装到微孔MOF 中的这种设计理念在以后的工业催化剂开发中起到了指导作用。反应机理如下:Co基MOFs 吸附了CO2分子,然后在活性金属中心(Pt 纳米颗粒和Au@Pd 纳米颗粒)上裂解CO2的C=O 键形成CO。稳定性实验表明,Pt/Au@Pd@UiO-66 可以在30 h 以上保持催化活性。由于高的氢化活性,负载在UiO-66 上的Pt 纳米颗粒对CO2加氢是有益的,并且核中的Au@Pd 纳米颗粒可以防止纳米颗粒聚集。

图2 (a)Pt/Au@Pd@UiO-66 纳米复合材料的合成过程;(b)Pt/Au@Pd@UiO-66 在不同温度下催化CO2 还原;(c)Au@Pd@UiO-66 和Pt/Au@Pd@UiO-66 的CO 选择性[30]Figure 2 (a): Synthesis process of Pt/Au@Pd@UiO-66 nanocomposites; (b): CO2-reduction catalyzed by Pt/Au@Pd@UiO-66 at different temperatures; (c): CO product selectivity of Au@Pd@UiO-66 and Pt/Au@Pd@UiO-66[30]
Li 等[31]通过将ZIF-8 在氮气氛中焙烧得到含氮掺杂的炭材料,通过负载相同量的不同活性组分来催化二氧化碳加氢产生一氧化碳, 对比发现镍负载的催化剂对CO2加氢合成一氧化碳具有更高的催化活性,在320−420 ℃的反应温度下,CO2转化率随温度的升高逐渐上升,最高达到了45%,CO 选择性基本维持在100%(图3)。这是由于ZIF-8 衍生的吡啶氮和碳化物对CO2进行吸附,高度分散镍的碳化物和金属物种为反应提供活性位点。Gutterød 等[32]通过将Pt 与联吡啶基连接,将Pt 导入Zr-UiO-67 中,得到Pt 高度分散的催化剂。在环境大气压,220−280 ℃,接触时间τ= 0.004−0.01 gcat/(min·mL),H2/CO2= 0.2−9.0 下,在固定床装置中评估了CO2氢化为CO,以确定RWGS 反应过程中MOF 晶格的影响及其稳定性。实验结果表明,这些催化剂在60 h 内具有很高的稳定性,一氧化碳选择性均大于90%,并且活性一直保持稳定,CO2转化率与Pt 的还原度成正相关。此外,在220−280 ℃进行了接触时间变化测试,以确定CO和CH4的形成顺序。观察到CO 的转化率和CO2转化率之间线性相关,而CH4的转化率相对较低,并且转化率高于零,表明CH4是CO 形成的二次产物。

图3 (a)M/ZIF-8-C 的CO2 转化率;(b)CO、CH4 的选择性[31]Figure 3 (a): CO2 conversion; (b): selectivity of CO, and CH4 on M/ZIF-8-C[31]
1.2 CO2 加氢制甲烷
二氧化碳的甲烷化反应也称为萨巴蒂埃反应(CO2+ 4H2→ CH4+ 2H2O,ΔH298= −165.9 kJ/molΔG298= −130.8 kJ/mol)[33]。从热力学角度看,CO2甲烷化反应在高温时受热力学平衡的限制,低温时更有利于正反应。从动力学角度看,CO2甲烷化反应在较低温度下反应速率较低,反应周期较长。因此,探索新型高活性催化剂是提高该反应收率的关键。一般来说,Ni、Co、Ru、Rh、Pd 等金属作为各种载体上的活性成分,在CO2甲烷化反应中都有活性[34−36],然而,目前MOFs 作为CO2甲烷化催化剂的研究尚处于起步阶段,相关报道较少。事实证明,MOF 具有有限的孔隙空间,可以作为潜在的载体,以防止金属以纳米颗粒形式迁移和聚集,从而确保催化剂的高活性。Lin 等[37]通过将Ni 纳米粒子限制在空心MOF 基衍生物中合成了一种新型的催化CO2甲烷化的材料(Ni@C),该材料呈现出空心球状结构。在325 ℃下,CO2和CH4的转化率都达到100%,连续反应24 h 后,Ni@C 杂化物可以仍能保持催化活性。作者提出了另一种通过CO2-TPD-MS 测量的反应途径,即CO2分子被直接氢化生成甲烷。提出了在Ni@C催化剂上可能的CO2甲烷化机理:首先,Ni@C 杂化体分别吸附CO2和H2进行活化和离解。形成表面中间体“Ni-CO2”,然后,“Ni-CO2”与解离H 原子相互作用形成“Ni-C-(OH)2”中间体, 最后,“ Ni-C-(OH)2”与解离的H 原子反应生成CH4和H2O。Zhen 等[38]以MOF-5 为载体,采用浸渍法制备了(x= 5、7.5、10、12.5)Ni@MOF-5 系列催化剂。所制备的催化剂在CO2甲烷化反应中表现出了良好的低温活性和稳定性。在MOF-5 上均匀分散Ni 颗粒以促进CO2甲烷化,结果表明,在320 ℃,H2/CO2= 4, GHSV = 2000 h−1,压力0.1 MPa 条件下,含镍10% 的Ni@MOF-5 的CO2转化率为75.09%,CH4选择性为100%,表明MOF-5 是制备CO2甲烷化催化剂的理想载体。作者认为在MOF-5 上高度分散的Ni 颗粒可以促进氢化反应的进行,随着Ni 负载量的增加,CO2的转化率增加,但是由于Ni 颗粒的表面分离,过量的Ni 负载量阻碍了反应。此外,Ni@MOF-5 在甲烷化反应的80 h 内表现出良好的稳定性,可能的机理是将CO2还原为CO,然后氢化生成甲烷。随后,他们课题组通过双溶剂法(DSM)和多次浸渍法(IM)将Ni 颗粒封装到高度有序的MIL-101 中,从而在低温下将CO2加氢生成甲烷[39](图4)。结果表明,在相同条件下,20%的Ni@MIL-101(DSM) 比20% 的Ni@MIL-101(IM)表现出更高的催化活性和稳定性(甲烷的转换频率在300 ℃下为1.63 × 10−3s–1)以及更低的活化能(88.01 kJ/mol)。作者发现Ni NPs 的形态和尺寸在高活性CO2甲烷化中起着重要作用,而超小(2.6 nm)、高度分散(42.3%) 的Ni NPs 则具有更大的作用。固定在MIL-101 框架中的裸露Ni(111) 面在高活性CO2甲烷化中也起着关键作用。此外,DFT 计算 结 果 表 明,(111) 面 暴 露 的Ni NPs 比 暴 露的Ni(111)面更具有CO2甲烷化活性。对20Ni@MIL-101(DSM)催化该过程的反应机理进行了如下解释:首先,CO2被化学吸附在催化剂上,并在表面分解为氧(Oads)和碳(COads)。其次,通过氢化过程将COads进一步转化为CHOads物质,然后形成CHx物质,如CH4。这些发现对未来低温区高效镍基CO2甲烷化催化剂的开发具有一定的指导意义。通过H2-TPR 分析,Ni@MIL-101(DSM)的高活性归因于Ni 颗粒与MIL-101 更强的金属-载体相互作用。Kennedy 小组[40]将Ru 颗粒引入UiO-66,该催化剂是单斜晶和四方ZrO2的混合物,并含有Ru纳米颗粒,在最优化反应条件下,负载1 %Ru 的UiO-66 上CH4的选择性为99%(图5)。该工作为通过MOFs 模板原位合成高效催化剂提供了新的方向,而无需进行高温煅烧或化学蚀刻处理。

图4 (a)xNi@MIL-101(DSM)合成示意图;(b)CO2 甲烷化在20Ni@MIL-101(DSM)催化剂上的反应机理[39]Figure 4 (a): Schematic illustrationonthesynthesisofxNi@MIL-101(DSM) catalysts;(b):The proposed possible reaction mechanism of CO2methanationover 20Ni@MIL-101(DSM) catalyst[39]

图5 Ru/UiO-66合成和原位转化为MOF 衍生催化剂的示意图[40]Figure 5 Sc hemeof synthesisand in situ transformation Ru/UiO-66 to MOF derived catalyst[40]
Li 等[41]选择ZIF-67 作为模板获得Co 基多孔炭材料以催化CO2甲烷化,在合成过程中加入不同含量的十六烷基三甲基溴化铵(CTAB)作为表面活性剂来调控ZIF-67 的晶体尺寸和形貌。将其原始粒径精确地从100 nm 调整为1 μm,并且形态可以从截形立方变为菱形的十二面体。炭化后,随着多孔炭材料形态从菱形十二面体变为立方体,0.01%的十六烷基三甲基溴化铵的最大微孔体积为0.125 cm3/g。在270 ℃、72000 m L·g–1·h–1、添加0.01% 十六烷基三甲基溴化铵的催化剂的CO2转化率为52.5%、甲烷选择性为99.2%(图6)。这项工作提供了一种有效调节金属纳米颗粒的形态和尺寸的方法,并成功地阻止了它们的烧结,这也为低温CO2甲烷化提供了良好的前景。
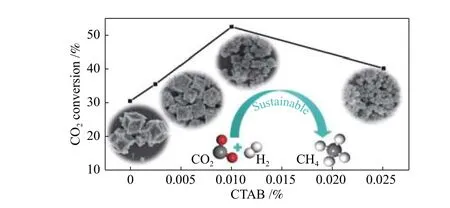
图6 添加不同含量十六烷基三甲基溴化铵的催化剂的CO2 转化率[41]Figure 6 CO2 conversion on catalysts with different content of CATB[41] (w ith perm ission from ACS)
1.3 CO2 加氢制甲酸
甲酸(HCOOH)是一种有价值的碱性化学品,通常在各种行业中用作防腐剂、除冰剂、杀虫剂以及抗菌剂。此外,它是一种很有前途的储氢材料[42,43]。然而,CO2加氢合成甲酸的主要问题是其热力学和动力学局限性,因此,人们期望开发一种有效的催化体系。该反应最常用的催化剂类型是:异质化分子催化剂、无载体和载体的纳米金属或本体催化剂。Maihom 等[44]利用DFT 计算发现,Cu-MOF-5 是CO2加氢合成甲酸很好的候选材料,并且反应的活性与催化剂的电荷转移有很大关系,电荷转移可以促进CO2活性,活化所需的能量随着电子基团的取代而减少,同时还有助于氢分子H−H 键的断裂。选用铜醇盐修饰的MOF-5 作为模型,以M 06-L 密度泛函法计算CO2加氢生成HCOOH 的反应过程,反应可以通过两种不同的途径进行,即协同机理和逐步机理。在协同机理中,反应在没有反应中间体的情况下直接完成的,它需要活化能为67.2 kcal/mol;而更受人们青睐的逐步机理则经历了两个步骤,包括形成甲酸酯中间体并将该中间体转化为HCOOH,第一步和第二步的活化能比较底,分别为24.2 和18.3 kcal/mol。此外,还对气相未催化反应进行了研究,以突出Cu-MOF-5 系统的催化作用,对于气相未催化反应计算得出的反应势垒为73.0 kcal/mol,明显高于Cu-MOF-5 系统的势垒(图7)。这些结果表明,金属官能化的MOF 可以用作将CO2加氢成HCOOH 的特殊催化剂,并且它们还可以稳定氢化反应系统中的所有物质。Johnson 小组[45]通过DFT 计算分析了UiO-66-P-BF2系统用于CO2加氢的过程,将受挫的路易斯(LPs)对引入UiO-66 后,所得的UiO-66-P-BF2可以有效地与CO2结合并解离H2,这将促进该反应过程(图8)。Johnson 再次使用DFT 模拟和筛查了一系列官能团,将这些官能团连接到UiO-66晶胞中的一个BDC 接头上,用于将CO2加氢成HCOOH。由于低的势垒,UiO-66-P-BF2分两步催化了该反应:H2首先在Lewis 对上解离(H2(vdW)→2H*((vdW)表示范德华络合物),然后氢化的氢与CO2的C 原子反应,质子氢与CO2的O 反应形成HCOOH(CO2+ 2H*→HCOOH)。计算表明,UiO-66-X将CO2加氢成HCOOH 的能垒按以下顺序减小:PB(NO2)2> P-B(CF3)2> P-B(CN)2> P-BBr2> PBCl2> P-BH2> P-BF2 > P-B(CH3)2。进一步计算分析了由R取代基上的八个不同官能团修饰的UiO-66-P-BF2材料的能级、势垒和几何形状,用于CO2加氢生成HCOOH[46]。最近,Ma 及其同事[47]合成了一系列Ru 锚定NH2-MIL-101(Cr) 材料,用于CO2加氢合成HCOOH,在120 ℃、6 MPa、CO2/H2=1∶1 时,转化数(TON)达到831,表明它们可以作为有效的CO2加氢制甲酸催化剂。Zhang 等[48]制备了一种新的镧(Ⅲ)MOF(JMS-1a)并对其进行功能化以获得Ru(Ⅱ)@JMS-1a 催化剂,在110 ℃、5.0 MPa、CO2/H2= 1∶4,24 h,5 mmol KOH 下,甲 酸 产率为98%。这是第一次报道La(III)MOF 可以作为将CO2转化为甲酸的催化剂。
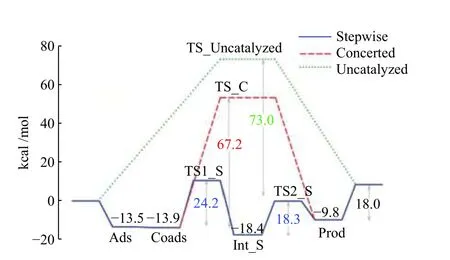
图7 协同和逐步机理的CO2 加氢能谱[44]Figure 7 Energy profile of the CO2 hydrogenation for concerted and stepwise systems[44]

图8 (a)LPs 功能化UiO-66 的八面体笼的BDC 配体(b)UiO-66-X 中CO2 加氢的反应能垒与H2 在UiO-66-X 中的吸附能的关系[45]Figure 8 (a): LPs functionalized BDC ligand of the octahedral cage of UiO-66; (b): Calculated reaction energy barriers for CO2 hydrogenation in UiO-66-X as a function of the adsorption energies of H2 in UiO-66-X[45]
1.4 CO2 加氢制甲醇
甲醇是用于各种有机合成的另一种C1 资源,可以用作化石燃料,燃料添加剂和生产多种化学品的前体的中间品,CO2加氢合成甲醇是保护环境的可行策略。CO2加氢系统需要高稳定性和高活性的材料作为催化剂来促进反应,因此,MOF作为一种新型多相催化剂对于二氧化碳的活化及其加氢制甲醇和回收释放的二氧化碳以及发展甲醇经济具有非凡的意义。Tshuma 等[49]用ZIF-8 作为Zn 源,用“酸性蚀刻-自组装”的方法,合成了CuZn 双金属修饰的MOFs,然后在450 ℃焙烧4 h得到了Cu/ZnO 催化剂(图9)。该催化剂不仅解决了MOFs 材料浸渍法活性组分负载量较少的问题,而且较传统的共沉淀方法制备的Cu/ZnO 催化剂粒径更小、金属分散更加均匀、保留了一定的MOFs 前驱体的形貌。在CO2加氢合成甲醇的反应中,反应活性好,甲醇选择性更高,并且在220−260 ℃,甲醇的选择性几乎没有降低,在275 h 的连续反应中,催化剂的活性维持稳定。
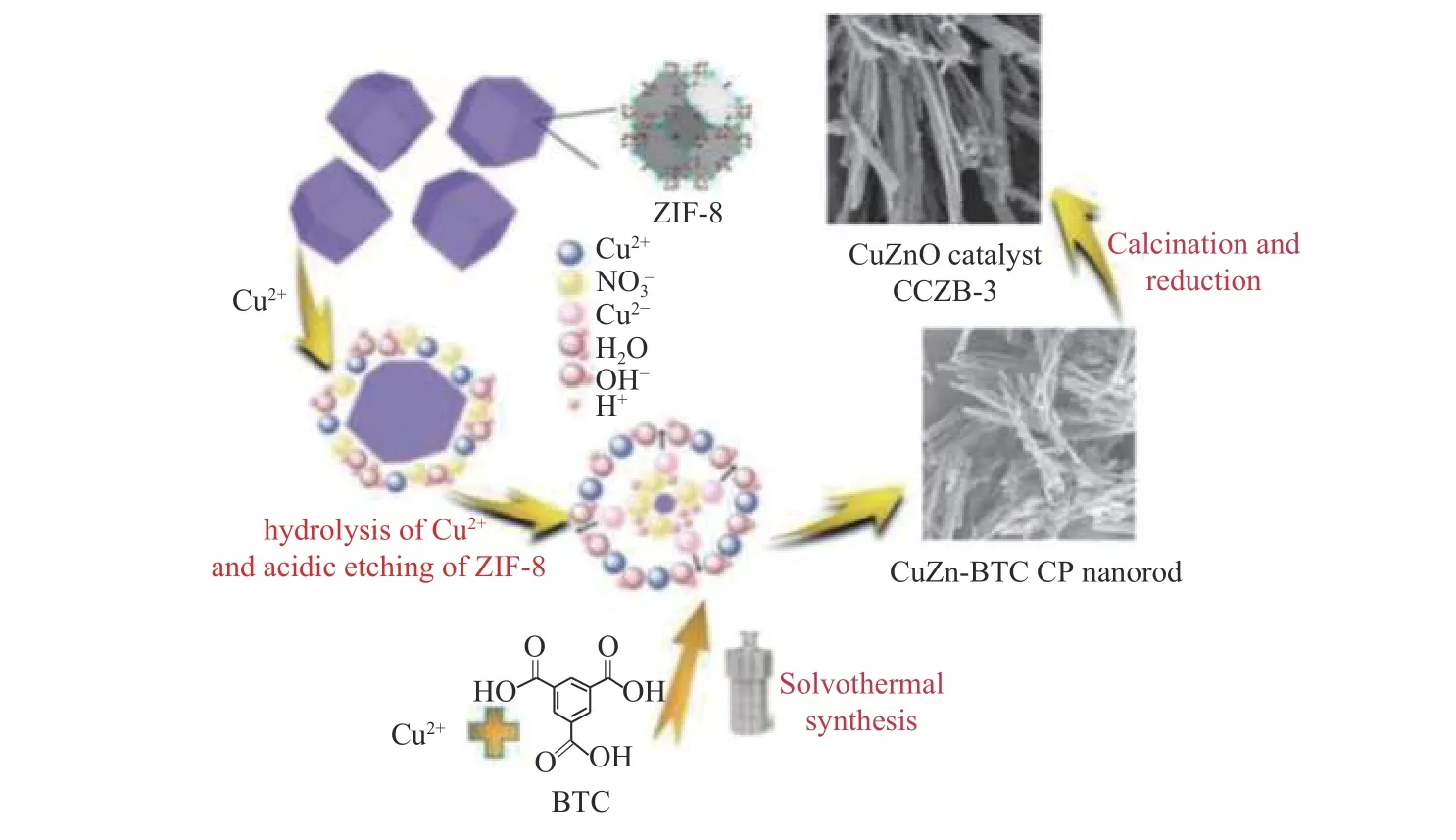
图9 以ZIF-8为牺牲模板制备CuZn-BTC的形成机理示意图[49]Figure 9 Schematic diagram oftheformationmechanismofCuZn-BTCpreparedwithZIF-8as a sacrificial template[49]
Rungtaweevoranit 等[50]在2016 年报道了一项开创性工作,使用UiO-66,Zr6O4(OH)4(BDC)6(BDC =1,4-苯二甲酸) 作为多孔基质来封装18 nm Cu 纳米晶体(NCs) 生成单晶 Cu ⊂UiO-66用于CO2加氢制甲醇(图10)。他们在175 ℃和1.0 MPa 下使用7 cm3/min 的CO2和21 cm3/min H2的气体混合物进行反应,以测试不同的SBU 和传统载体与Cu NCs对CO2加氢催化性能的影响。结果表明,只有ZrO2上 的Cu,UiO-66 上 的Cu,Cu/ ZnO/Al2O3和Cu ⊂UiO-66 表现出催化活性,而 Cu ⊂UiO-66上甲醇形成的转换频率(TOF)最高(3.7 × 10–3s–1)。而MIL-101(Cr)和 Cu ⊂ZIF-8上的Cu不能将CO2催化转化为甲醇,表明在该反应中只有Zr 或Zn氧化物保持了Cu NCs 的性能, Cu ⊂UiO-66随温度升高而形成的CO 的初始TOF 总是高于以Cu/ZnO/Al2O3为基准的催化剂,在所有反应温度下, Cu ⊂UiO-66上未检测到CO。与Cu/ZnO/Al2O3相比,在175 ℃下,Cu ⊂UiO-66的收率稳定提高了8 倍,甲醇选择性达到了100%。此外,XPS分析证明, Cu ⊂UiO-66的高选择性和出色的活性是由于Cu NCs 和UiO-66 的Zr-SBU(MOFs 的纳米级金属氧化物的二级结构单元)之间的强相互作用。有序的精确排列在Cu 表面上,Cu 和Zr-SBU [Zr6O4(OH)4(-CO2)12]之 间具 有良好接触。因此,Rungtaweevoranit 等假定Cu NCs和Zr 氧化物SBU 之间的界面是Cu NC-UiO-66 催化剂的活性位点。这为开发具有选择性高、效率高的基于MOF 的多相催化剂催化加氢提供了新的见解。这也是第一次发现MOFs 中的SBUs 可以像金属氧化物一样与载体有强烈的相互作用。为了进一步设计和解释用于CO2加氢的基于MOF的特定且稳定的表面界面结构催化剂,An 等[51]使用 的UiO-bpy(bpy = 2,2'-联 吡 啶)MOF 包 含Zr6SBUs {Zr6(μ3-O)4(μ3-OH)4}作为载体,并且Cu(II)离子已预先组装在bpy 位置和 MOF 中Zr6团簇上修饰的Zn(II) 离子在250 ℃和4 MPa,H2/CO2= 3 的条件下原位还原合成金属化的UiO-bpy 生成超小的Cu/ZnOx纳米颗粒(图11)。获得的UiO-bpy MOF 可以锚固生成的超小的Cu/ZnOxNPs(纳米颗粒),有效地将Cu NPs 与ZnOx分离,将其限制在MOF 腔中,并防止Cu NPs 团聚。用后合成的方法将Cu 和Zn 负 载 在Zr 基MOFs材 料UiO-bpy 上,获得的Cu/ZnOx@MOF 在CO2加氢过程中表现出很高的催化活性,该催化剂的甲醇时空收率达到2.59 g·kgCu–1·h–1,是商业化Cu/ZnO/Al2O3催化剂的3 倍,在100 h 内还具有100%的甲醇选择性和良好的稳定性。此外,在通过XRD(PXRD)、N2吸附-脱附、等温曲线、TEM、漫反射UV-vis-NIR 光谱分析等方法对催化剂结构表征后,他们发现原位形成了超小Cu/ZnOxNPs 直径小于2 nm并被高度分散在MOF 的纳米腔中,因此,Cu/ZnOx和Cu/Zr6簇界面处的Cu/Zn/Zr 原子占所有Cu/Zn/Zr 的50%以上,可用于研究催化活性界面。此外,在反应条件(H2/CO2= 3,250 ℃,0.1 MPa)下,对MOF SBU 和Cu/ZnOx物种进行XPS 分析,以研究Zn、Cu 和Zr价态的变化。分析结果证实,在催化过程中,部分Zr(IV)和Zn(II) 由于Cu 表面游离的氢溢出到Zr基SBU 中而被还原为Zr(III) 和Zn(0)的低价态。他们通过金属纳米颗粒与有机螯合配体和MOF的SBUs 之间的特定且可调节的强金属-载体相互作用,增加了使用官能化MOF 代替传统金属氧化物作为新型载体的知识。与共沉淀方法制备的Cu/Zn/ZrO2催化剂[52]相比,无论是醇的选择性,还是产率,都有很大的提升。因此,它为优化和设计基于MOF 的高活性催化材料提供了新思路。
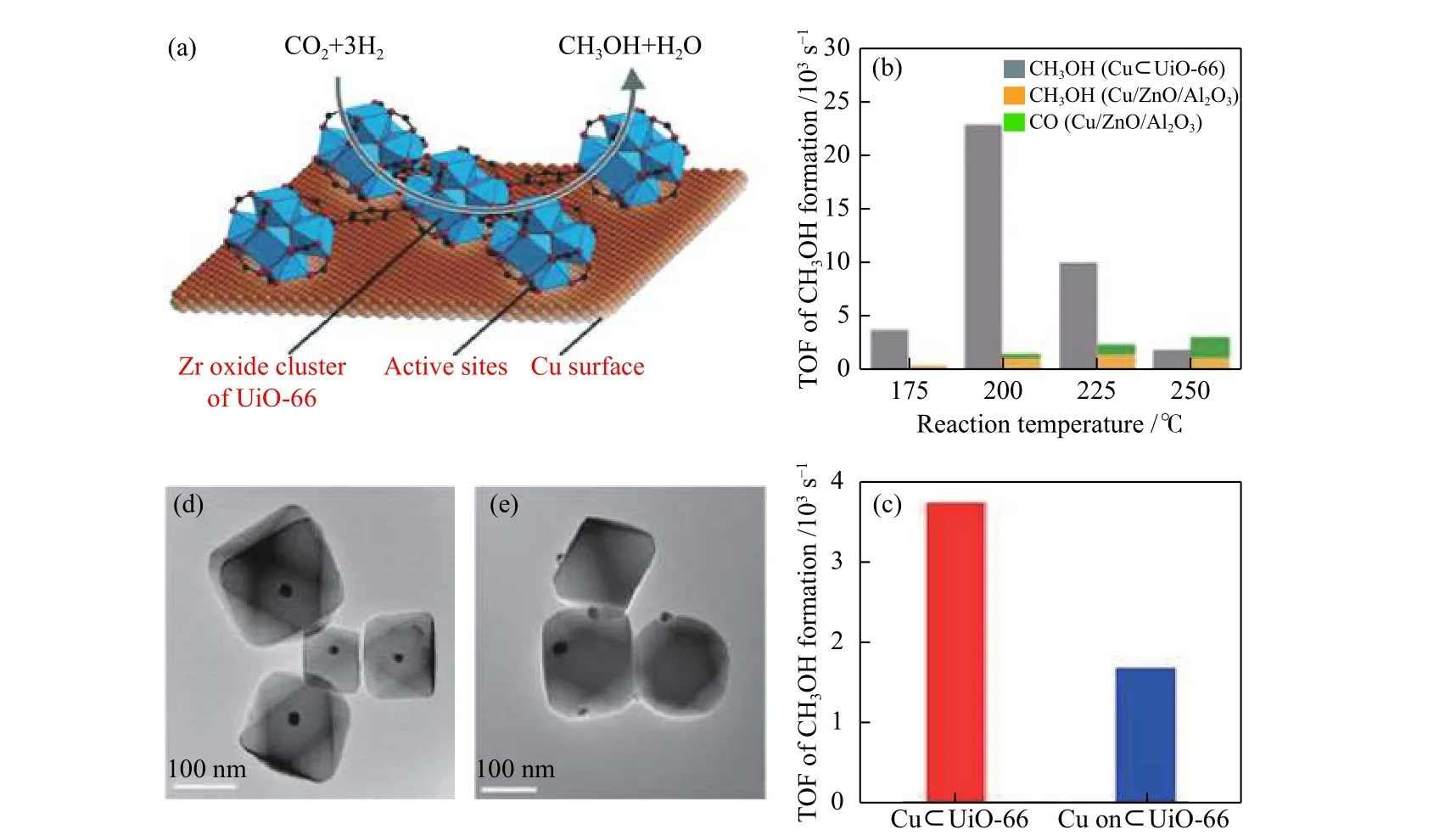
图10 (a) C u ⊂UiO-66活 性位点的图示;(b)C u ⊂UiO-66和 Cu/ZnO/Al2O3 在各不同反应温度下产物的TOFs;(c)在Cu ⊂UiO-66上和Cu 在UiO-66 上甲醇的初始TOFs;(d)C u ⊂UiO-66(UiO-66 内部的单个CuNC)的TEM;(e)UiO-66 上Cu 的TEM[50]Figure 10 (a) Illustration of active site of C u ⊂UiO-66 catalyst; (b) TOFs of product formation over C u ⊂UiO-66 catalyst and Cu/ZnO/Al2O3 catalyst as various reaction temperatures; (c) Initial TOFs of methanol formation over C u ⊂UiO-66 and Cu on UiO-66 ; (d) TEM images of C u ⊂UiO-66(single Cu NC inside UiO-66); (e) TEM images of Cu on UiO-66[50]

图11 (a)合成后金属化的UiO-bpy 原位还原制备CuZn@UiO-bpy 示意图;(b)CH3OH 的时空产率随在反应时间的变化;(c)产物的选择性随反应时间的变化;(d)MOF 中封装的活性位点以及各种表面位点在催化CO2 加氢中的功能[51]Figure 11 (a): Preparation of CuZn@UiO-bpy via in situ reduction of post-synthetically metalized UiO-bpy; (b): STY of CH3OH vs reaction time on stream; (c) Selectivity of product vs reaction time; (d) Schematic showing the encapsulated active sites inMOFandthefunctionsofthe various surfacesites in catalytic CO2hydrogenation[51](withpermission from ACS)
贵金属催化剂研究最多的是Pd 基催化剂,Pd 在高温情况下可以和Zn 形成PdZn 合金,这也是CO2加氢合成甲醇的活性位,因为它可以有效地抑制逆水煤气反应[53]。此外,高分散度的金属纳米颗粒、小粒径的PdZn 合金对于甲醇的选择性和产率是至关重要的,MOFs 材料可以更好地实现这点。Ye 等[54]通过直接煅烧Pd@ZIF-8 前体将CO2加氢成甲醇,开创了一种新型的高效PdZn 合金催化剂的制备方法。首先,考虑到MOF 的限制作用的优势,通过将超小的Pd 颗粒嵌入ZIF-8 的骨架孔中来合成Pd@ZIF-8,然后将获得的Pd@ZIF-8 在不同温度(350、400 和500 ℃)下,在空气中热解,制备了催化剂PZ8-t(t代表热解温度)。因此,在生成的PZ8-t中形成一定数量的具有强金属-载体相互作用(SMSI)的Pd-ZnO 界面。TEM 分析显示Pd 颗粒处于亚纳米级,在ZIF-8 基质中的平均尺寸为(1.2 ± 0.2)nm。同样,在预还原过程中,这些亚纳米级的Pd 颗粒倾向于转变为PdZn 合金相。ZIF-8 的孔隙结构可以限制Pd 颗粒的团聚生长,同时高比表面积的多孔ZnO 也确保了Pd 纳米颗粒的高度分散,将PZ8-t热解并通过H2预还原,金属Pd 转化为PdZn 合金。通过在空气气氛下焙烧得到Pd/ZnO 催化剂,用于将CO2转化为甲醇(图12)。在4.5 MPa,250−290 ℃和 H2/CO2= 3 的条件下,催化测试结果表明,在270 ℃,TOF 为972 h−1下PZ8-400 的甲醇收率最高达到0.65 g/(gcat·h),反应50 h 后,甲醇的选择性从51.9%逐渐增加到54.2%,而CO2的转化率从15.1% 下降到13.6%。高催化活性归因于小尺寸的PdZn 合金颗粒以及ZnO 表面的氧缺陷。此外,Pd 和ZnO 之间的相互作用也使Pd-ZnO 合金催化剂具有较长的耐久性。通常,理论计算在新材料的探索中起着重要作用。Johnson小组[53]通过DFT 模拟计算分析了UiO-67(NBF2)4的催化活性,将受挫的路易斯对(FLPs)引入UiO-67 的配体后,新催化剂具有路易斯酸碱位,可以捕获CO2并将其转化为甲醇。计算结果表明,具有最低势能面的反应路径如下:CO2→cisHCOOH→CH2(OH)2→CH2O→CH3OH。
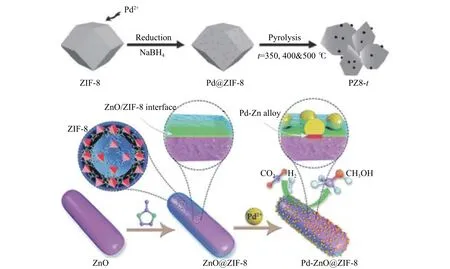
图12 PZ8-t 催化剂的制备和Pd-ZnO@ZIF-8 催化CO2 加氢制甲醇的示意图[54]Figure 12 Schematic illustration of the preparationprocessof thePZ8-T and thepreparation procedure of Pd-ZnO@ZIF-8 forCO2 hydrogenationto methanol[54]
1.5 CO2 加氢制烯烃
许多研究表明,铁基络合物对二氧化碳加氢制备烯烃具有很高的催化活性[55]。因此,可选择MOFs 作为模板来引入铁基络合物,以构建这种转化反应的新型高效催化剂。Hu 等[56]将α-Fe2O3引入MIL-53 或ZIF-8,以获得用于CO2加氢的新催化剂。在不同的溶剂体系中进行合成,获得了具有不同尺寸的ZIF-8 以装载α-Fe2O3, 在p= 3 MPa、t= 300 ℃时,MIL-53 表 现 出 较 高 的CH4选 择 性,对轻烯烃没有选择性,而轻烯烃的选择性随ZIF-8尺寸的增大而降低。作者认为,高催化活性归因于CO2而不是CO 的高吸附能力,ZIF-8 的高H2吸附能力可富集H2以生成烯烃。随后,他们的小组通过一步法热解Fe-MIL-88B 和热解铁改性的ZIF-8以形成带有金属颗粒的N 掺杂空心球,合成了有效的CO2加氢催化剂,以得到各种碳氢化合物[57]。
MOFs 富含C、N 和金属元素,因此一些Fe 基的MOFs 材料受到了关注。Liu 等[58]以Fe-MIL-88B为前驱体, 通过控制在氮气中的焙烧条件,发现N-600-0 催化剂效果最好,CO2的转化率最高可达46.0%,C5+和C2−C4烯烃的选择性分别为22.2% 和19.2%,(图13)。

图13 在不同催化剂上的二氧化碳转化率和产物选择性[58]Figure 13 CO2 conversion rate and selectivity on different catalysts[58] (with permission from Elsevier)
Ramirez 等[59]选 择Fe 基MOF Basolite F300作为模板来煅烧以形成Fe/C 杂化材料(图14)。在Fe/C 杂化材料中添加K+可以提高加氢反应过程中烯烃的选择性。在p= 3 MPa、t= 350 ℃、H2/CO2= 3、空 速24000 mL·g–1·h–1和Fe/(C + K) =0.75 的条件下,催化剂可以有效地促进CO2加氢转化,C2−C6烯烃的选择性为40%,C2−C6烯烃在产物中的选择性最高。催化剂具有良好催化活性的可能原因是K+的强碱度和电子给体可以促进CO2接受来自Fe/C 的电子以增强Fe-C 相互作用。由Basolite F300 制备的炭材料催化剂,性能超过了传统材料的催化剂,有很好的工业化前景。
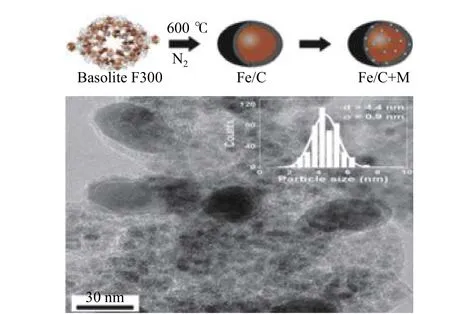
图14 Fe 基Basolite F300 催化剂合成机理与形态表征[59]Figure 14Synthesismechanism and morphologyofFe-based BasoliteF300[59] (withpermission from ACS)
2 MOFs 在CO2 加氢中的优点和存在的问题
2.1 优点
基于上述分析MOFs 材料在CO2加氢反应中具有以下特点:(1)能够通过自身的孔道结构和大比表面积限制活性组分的聚集和生长并使其均匀的分散;(2) 可以直接为反应提供催化活性中心,也可以作为载体将具有催化活性的物质连接或负载到MOFs 材料上;(3)具有开放的骨架和规整的孔道结构,可作为可官能化的催化剂载体;(4)三维框架形成独特的孔或通道并显示出较大的比表面积,可富集二氧化碳并提供足够的反应空间。另外,一些催化活性材料可以装在大孔中,以增强基于MOFs 的材料的催化性能;(5)某些基于MOFs的材料具有很高的化学稳定性和热稳定性,这为CO2转化奠定了基础。已经证实了Lewis 功能化的MOFs 用于CO2加氢的低势垒途径。
MOF 基催化剂可以获得CO2加氢制甲醇的高选择性。Muller 等[60]将Cu 和ZnO 纳米颗粒负载到MOF-5 上,考察了CO/CO2/H2混合气合成甲醇的催化性能,相比传统Cu/ZnO 催化剂的活性有很大的提高,甲醇产率提高一倍以上。Wang 团队[51]利用UiO-bpy MOF(bpy 为2, 2'-联吡啶)锚定Cu/ZnOx纳米粒子,有效防止Cu 纳米粒子团聚,以及Cu 纳米粒子与ZnO 之间的相互分离。该催化剂的时空产率达2.59 g·kg–1·h–1,比商业Cu/ZnO/Al2O3催化剂催化活性高出3 倍;同时该催化剂也具有100%的甲醇选择性和良好的稳定性。以功能化的MOFs 取代传统金属氧化物作为载体,具有催化活性的金属纳米粒子与有机螯合配体及金属氧簇节点之间的强金属-载体相互作用,优化了催化活性和选择性。
2.2 存在的问题
MOFs 作为一种新颖的催化剂载体,利用其孔道结构的限域功能,使超细活性组分均匀地分散在孔道中。所制备的催化剂一般稳定性好,产物选择性高,所以MOFs 受到越来越多的关注,在很多重要的催化转化中已经显示出巨大的应用潜力,但目前该领域的研究还处于起步阶段,仍有许多问题亟待解决。提高CO2加氢的稳定性,活性和产物收率,将CO2用于MOF 上的非均相C−H活化和羧化反应,使用CO2作为与氧化物或烯烃的一步共聚反应的单体。作为催化剂或催化剂载体,MOFs 需具有高稳定性,至少在反应状态下保持稳定。然而MOFs 中金属离子和有机配体是通过弱的配位键自组装而成的,MOFs 稳定性通常较差(尤其在水溶液中可能更差),因此,今后要着力于高稳定性MOFs 的设计合成。Zhen 等[61]通过浸渍法制备了10NiMOF-5 催化剂,反应温度在320 ℃时,CO2转化率虽然达到75.09%,但是仅仅在100 h的测试中保持稳定的活性。Yazhi 等[62]合成的贵金属Pd/ZnO 催化剂,相较于传统的制备方法性能和稳定性上也有所提升。但是290 ℃下CO2最高转化率是18.4%,CO2的转化率偏低,这也是后续可以研究的方向。
用于二氧化碳加氢的MOF 催化剂的制备一般是先预合成MOFs 材料,然后通过各种方法(溶液浸渍、固相研磨、化学气相沉积、微波辐射等) 负载活性组分[63]。另一种方法主要是将预先制备的金属纳米颗粒加入到MOF 制备体系中来构建复合材料[50],这种方法可以控制MOFs 材料尺寸和形态,如常温条件下的直接包覆法和溶剂热法。研究人员还将MOFs 材料作为前驱体,在不同气氛下进行热解处理制备金属氧化物催化剂[29]。MOFs 的有机配位体将金属节点分开,使金属均匀分布在材料的整体骨架中,可以防止前驱体向氧化物转化过程中金属氧化物快速聚集产生的团聚现象。由于MOFs 材料的固有的微观结构,因此,难以充分引入金属前体进入MOF 的孔道,导致金属纳米颗粒沉积在MOF 的外表面。而且MOFs材料的制备并不容易,虽然已经有很多研究快速制备MOFs 材料的成果,大部分MOFs 的大量制备还是比较困难。Zhang 等[29]利用ZIF-8 为牺牲模板以1,3,5-苯三甲酸为有机配体,Cu2+、Zn2+作为金属离子合成了双金属MOFs 材料CuZn-BTC,并以此为前驱体,在空气中焙烧得到了CuO/ZnO 催化剂应用于CO2加氢制备甲醇,但该催化剂只保持了MOFs 前驱体的一定形貌。
对MOFs 包裹的金属纳米颗粒催化剂,当前的研究多集中于MOFs 的限域效应和孔道的尺寸效应,应该加强MOFs 与金属纳米颗粒的相互作用研究,这有望为高活性和高选择性的催化剂研制带来新的机遇。加强MOFs 的催化机理研究。尽管MOFs 基材料在一些反应中显示出优异的催化性能,但其催化机理还不清楚。Zr 基的MOF UiO-66 和UiO-bpy 催化剂,甲醇的选择性虽然都达到了100%,但是机理尚不明确,这可能是由于MOFs 催化剂可以得到的超小的高度分散的纳米活性组分导致。今后需运用同步辐射和原位表征技术等手段对反应状况下MOFs 进行详细表征和实时监控,以获得明确的催化反应机理,指导MOFs的选择设计合成。
3 结论与展望
综上所述,在过去十年中,由于MOF 材料独特的骨架结构,特别是其限制效应、功能和结构多样性,为CO2加氢催化剂的设计和制备提供了许多可能性。在使用MOF 及其衍生材料作为催化剂的CO2化学转化领域也取得了许多进展,特别是在CO2加氢和环加成反应方面。将CO2加氢为CH3OH、HCOOH、CO、CH4和其他所需化学物质被认为是减轻CO2排放的最有希望的方法之一。由于其限制效应和均匀分布的催化位点和功能化的晶体结构,MOFs 在CO2加氢反应中始终表现出出色的催化性能。从目前的研究来看,尽管MOF材料在该领域的探索才刚刚开始,但其出色的催化性能已展示出解决这一艰巨任务的希望。例如,在CO2加氢制甲醇方面,Cu/ZnOx@MOF 催化剂的产率最高为2.59 gCH3OH/(kgCu·h),甲醇的选择性为100%,超过了商用催化剂Cu/ZnO/Al2O3,ZnO-ZrO2和In2O3/ZrO2。关于CO2→HCOOH,MOF(UiO-67)在索格利特型回流冷凝系统中用作固体分子催化剂,在大气压和85 ℃下获得了410 h−1的高周转频率。迄今为止,提高CO2加氢的稳定性、活性和产物收率仍然是一个艰巨的挑战。遗憾的是,与该领域的其他主要催化剂相比,许多原始MOF基材料的应用最终受到其苛刻条件下的稳定性的限制。传统的催化剂往往具有较高的CO2转化率,而MOF 催化剂很难实现。因此,应该在这一领域投入更多的精力来设计和合成新的MOF 材料。同时,有关CO2的活化及其加氢成芳族化合物,汽油和异链烷烃等MOF 基催化剂的报道极少。因此,MOF催化剂在提高CO2转化率和研究领域方面,仍有很多工作要做。尽管仍然存在许多挑战,并且笔者对本文所综述的多孔材料的理解仍然不完整,但是可以相信,具有特定性能和功能更强的MOF材料的设计和合成将在不久的将来会得到快速发展。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12 02:07:48
第一财经(2019年8期)2019-08-26 17:53:46
中国调味品(2017年2期)2017-03-20 16:18:13
哈尔滨医药(2015年2期)2015-12-01 03:57:13
学习月刊(2015年14期)2015-07-09 03:37:48
中学化学(2015年2期)2015-06-05 07:18:13
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
物理化学学报(2015年5期)2015-02-28 17:34:47
无机化学学报(2014年4期)2014-02-28 17:31:23

- 燃料化学学报的其它文章
- 生物质气再燃脱除流化床N2O 的机理研究
- Catalytic pyrolysis of sugarcane bagasse by zeolite catalyst for the production of multi-walled carbon nanotubes
- Nitrogen-doped porous carbon supported nickel nanoparticles as catalyst for catalytic hydroconversion of high-temperature coal tar
- Probing into the crystal plane effect on the reduction of α-Fe2O3 in CO by Operando Raman spectroscopy
- CoSOH/Co(OH)2 复合纳米片的制备及其氧析出催化性能
- Effects of promoters on carburized fused iron catalysts in Fischer-Tropsch synthesis