
HPLC-MS/MS法同时测定杞菊地黄丸(浓缩丸)中11种成分的含量
2021-11-18 09:19邓俊杰谢浙裕何丽娜
西北药学杂志 2021年5期
邓俊杰,谢浙裕*,何丽娜
(1.绍兴市食品药品检验研究院,绍兴 312000;2.浙江大学第一附属医院临床医学部,杭州 310000)
杞菊地黄丸(浓缩丸)现行标准为2020年版《中国药典》一部,处方由枸杞、菊花、熟地黄、酒萸肉、牡丹皮、山药、茯苓、泽泻8味中药制成,具有滋肾养肝的功效。用于治疗肝肾阴亏、眩晕耳鸣、羞明畏光、迎风流泪、视物昏花[1-2]。与传统剂型相比,浓缩丸具有服用量少、方便携带等优点。中成药的功效是多种活性成分共同作用的结果,但查阅《中国药典》和相关文献[3-7],仅有高效液相色谱(HPLC)法测定酒萸肉中莫诺苷、马钱苷和牡丹皮中丹皮酚含量的报道,缺少其余6味中药的质量控制指标,不能全面评价其整体质量。
根据2020年版《中国药典》和相关文献报道[8-11],枸杞的指标成分为甜菜碱;菊花的指标成分为绿原酸、木犀草苷和3,5-O-二咖啡酰基奎宁酸;熟地黄的指标成分为毛蕊花糖苷;山药的指标成分为尿囊素;茯苓的指标成分为茯苓酸;泽泻的指标成分为23-乙酰泽泻醇B。
本实验使用高效液相色谱-三重四级杆串联质谱(HPLC-MS/MS)法,以多反应监测模式进行检测,正负离子同时扫描,具有质谱灵敏度高、准确性好、选择性强的特点,建立了同时测定杞菊地黄丸(浓缩丸)中11种有效成分的方法。既简化了甜菜碱的提取方式,又解决了多成分液相检测时周期长、杂质干扰大的问题,对8味中药的各指标成分进行了含量测定,为杞菊地黄丸(浓缩丸)的质量标准制定提供了依据[12-16]。
1 仪器与试药
1.1仪器 1260-6470型高效液相-三重四级杆串联质谱仪(美国Agilent公司);XPE26百万分之一电子天平(瑞士METTLER TOLEDO公司);ZM-200型超离心碾磨仪(德国Retsch公司);FESCO17型微量离心机(美国Thermo Fisher公司);HH-4A型数显恒温水浴锅(常州国华电器有限公司)。
1.2试药 甜菜碱(批号110894-201604,质量分数为99.2%),绿原酸(批号110753-202018,质量分数为96.1%),木犀草苷(批号111720-201810,质量分数为93.5%),3,5-O-二咖啡酰基奎宁酸(批号111782-201807,质量分数为94.3%),毛蕊花糖苷(批号111530-201914,质量分数为95.2%),莫诺苷(批号111998-201703,质量分数为97.4%),马钱苷(批号111640-201808,质量分数为99.0%),丹皮酚(批号110708-201908,质量分数为99.8%),尿囊素(批号111501-200202,质量分数为100.0%),23-乙酰泽泻醇B(批号111846-201705,质量分数为99.7%),均购自中国食品药品检定研究院;茯苓酸(批号J2140009,质量分数为100.0%,ANPEL公司);甲醇和乙腈均为质谱纯,均购自Merck公司;甲酸(质谱纯,CNW公司);杞菊地黄丸(浓缩丸)(仲景宛西制药股份有限公司,批号:200511、200416、190806、190417)。
2 方法与结果
2.1液相色谱-质谱条件
2.1.1色谱条件 色谱柱:Agilent Porshell EC-C18(50 mm×4.6 mm,2.7 μm)柱;流动相:乙腈-1 mL·L-1甲酸(梯度洗脱,见表1),柱温:30 ℃,流速:0.5 mL·min-1,进样量:2 μL。
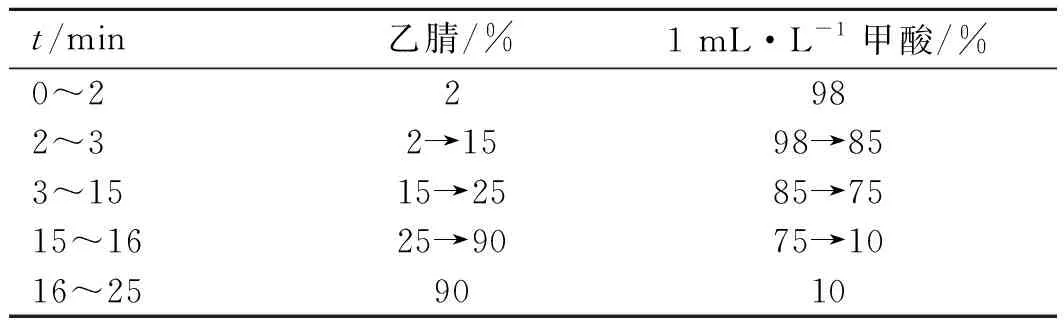
表1 梯度洗脱方法
2.1.2质谱条件 离子源:电喷雾离子源;干燥气和碰撞气:氮气(质量分数≥99.999%);干燥气温度:300 ℃;干燥气流速:6 L·min-1;扫描模式:正负离子同时扫描;毛细管电压:4 000 V(+),3 500 V(-);为提高检测灵敏度,采用分时间段的多反应监测(MRM)模式,电离模式、保留时间、定量离子对、碎裂电压和碰撞能量(CE),见表2。
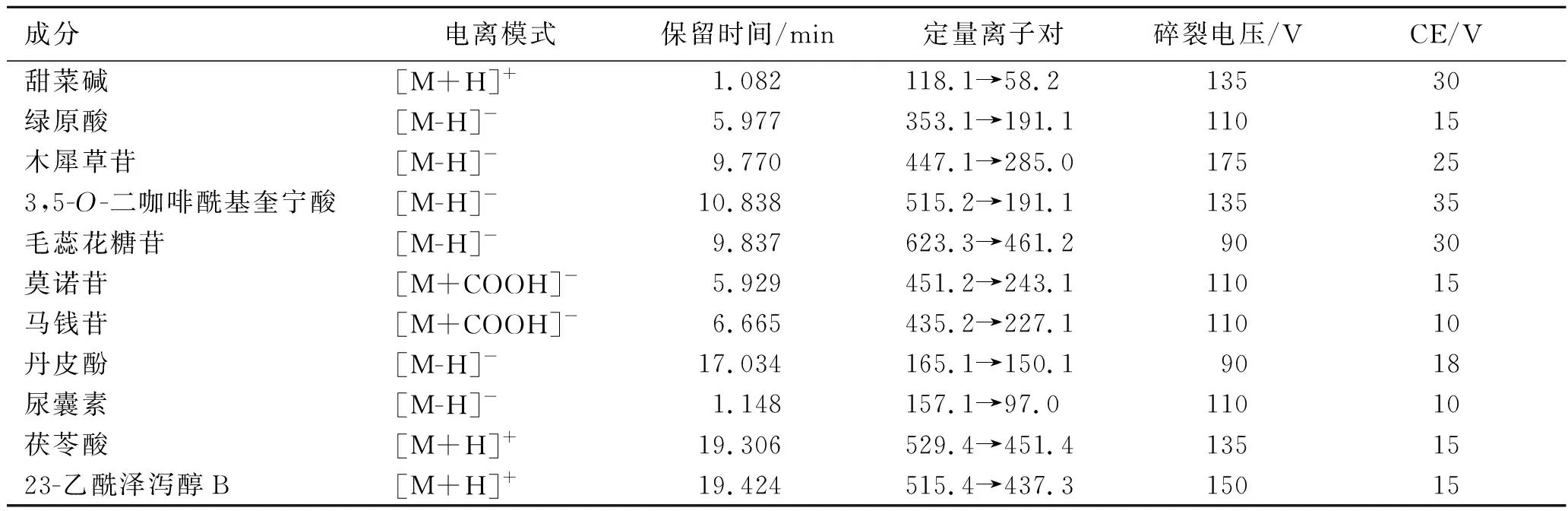
表2 11种成分的电离模式、保留时间、定量离子对、碎裂电压和碰撞能量
2.2溶液的制备
2.2.1对照品溶液 称取甜菜碱20.18 mg、绿原酸10.86 mg、木犀草苷10.98 mg、3,5-O-二咖啡酰基奎宁酸15.28 mg、毛蕊花糖苷15.79 mg、尿囊素10.12 mg、茯苓酸10.83 mg、23-乙酰泽泻醇B 15.65 mg,置于同一100 mL量瓶中,加入体积分数为70%的甲醇制得①号储备液;分别称取莫诺苷10.73 mg、马钱苷20.07 mg、丹皮酚25.95 mg,置于同一25 mL量瓶中,加入体积分数为70%的甲醇制得②号储备液。再精密量取①、②号储备液各2 mL,置于同一20 mL量瓶中,加入体积分数为70%的甲醇制成混合对照品溶液。
2.2.2供试品溶液 取本品适量(批号200511),研细,取2.0 g,精密称定,置于具塞锥形瓶中,精密加入体积分数为70%的甲醇50 mL,密塞,称定质量,加热回流1 h,取出,放冷,再称定质量,用体积分数为70%的甲醇补足减失的质量,摇匀,超高速离心,取上清液作为供试品溶液。
2.2.3阴性供试品溶液 按照处方组成及生产工艺制得缺枸杞、菊花、熟地黄、酒萸肉、牡丹皮、山药、茯苓和泽泻的阴性供试品,按照2.2.2项下方法制得阴性供试品溶液。
2.3方法学验证
2.3.1专属性考察 分别精密吸取混合对照品溶液、供试品溶液和阴性供试品溶液各2 μL,按照2.1项下色谱条件检测,记录质谱图。结果供试品显示出与对照品保留时间相同的质谱峰,阴性供试品溶液在此保留时间无质谱峰,其他组分对结果无干扰,表明该方法系统适用性良好。见图1。

图1 11种成分的提取离子流图
2.3.2线性关系考察 精密量取2.2.1项下制备的①、②号对照品储备液各2 mL,分别置于同一5、10、20、50、100 mL量瓶中,加体积分数为70%的甲醇稀释至刻度,制得系列混合对照品溶液。按照2.1项下色谱条件测定,以质量浓度为横坐标(x),峰面积为纵坐标(y),绘制标准曲线,计算得线性回归方程,结果11种成分在各自质量浓度范围内均呈良好线性关系。见表3。
2.3.3定量限 精密度量取2.2.1项下制备的混合对照品溶液,逐级稀释,按照2.1项下色谱条件测定,以信噪比为10∶1对应的质量浓度为定量限下限,结果见表3。
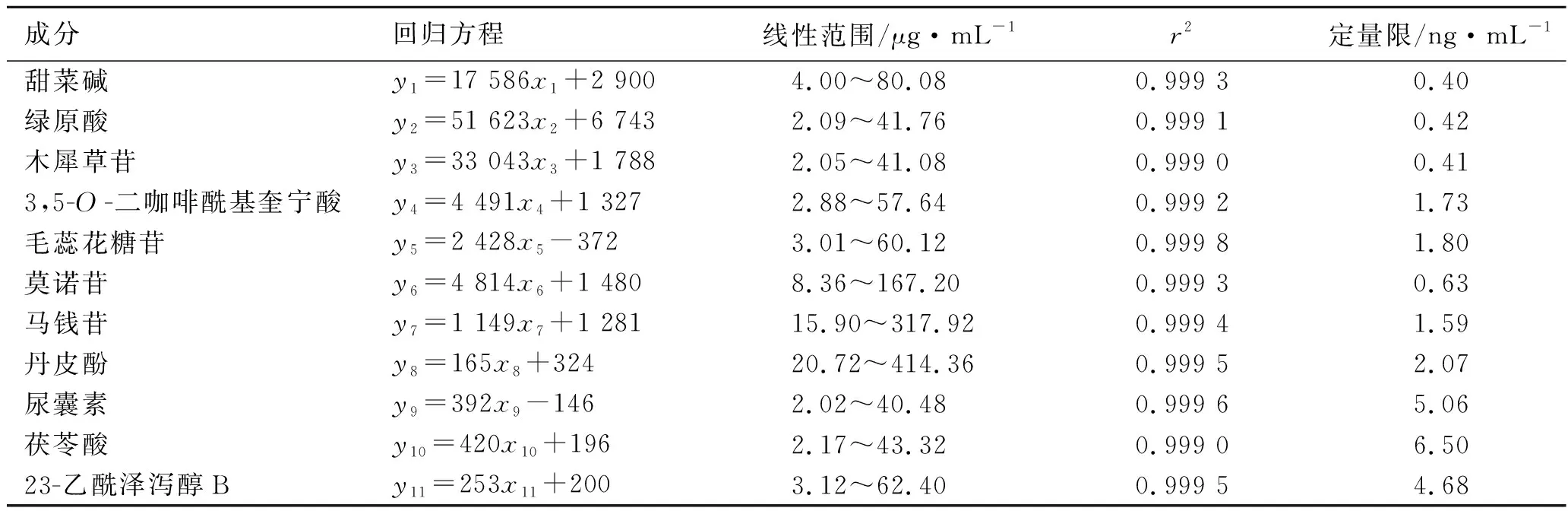
表3 11种成分的回归方程、线性范围、检测限和定量限
2.3.4精密度实验 精密量取2.2.1项下制备的混合对照品溶液,按照2.1项下色谱条件连续进样6次,结果甜菜碱、绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、毛蕊花糖苷、莫诺苷、马钱苷、丹皮酚、尿囊素、茯苓酸和23-乙酰泽泻醇B峰面积的RSD值分别为0.5%、0.7%、0.4%、0.9%、1.6%、0.8%、1.2%、0.9%、1.0%、1.4%、1.7%,表明仪器精密度良好。
2.3.5重复性实验 取6份同一样品粉末,按照2.2.2项下方法制备供试品溶液,按照2.1项下色谱条件测定,结果甜菜碱、绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、毛蕊花糖苷、莫诺苷、马钱苷、丹皮酚、尿囊素、茯苓酸和23-乙酰泽泻醇B峰面积的RSD值分别为0.7%、0.3%、0.5%、1.1%、1.7%、0.9%、1.9%、0.7%、1.5%、2.1%、1.3 %,表明该方法重复性良好。
2.3.6稳定性实验 取同一供试品溶液,分别在0、4、8、12、24、48 h,按照2.1项下色谱条件测定,结果甜菜碱、绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、毛蕊花糖苷、莫诺苷、马钱苷、丹皮酚、尿囊素、茯苓酸和23-乙酰泽泻醇B峰面积的RSD值分别为0.8%、0.7%、0.6%、1.4%、1.9%、0.7%、1.5%、1.2%、1.0%、1.7%、1.5%,表明稳定性良好。
2.3.7加样回收率实验 精密称取9份同一样品粉末(批号200511),每份1.0 g,分别置于具塞锥形瓶中,3份为1组,分别加入相当于1.0 g样品中各组分含量为80%、100%、120%的对照品,按照2.2.2项下方法制备供试品溶液,按照2.1项下色谱条件测定得甜菜碱、绿原酸、木犀草苷、3,5-O-二咖啡酰基奎宁酸、毛蕊花糖苷、莫诺苷、马钱苷、丹皮酚、尿囊素、茯苓酸和23-乙酰泽泻醇B的平均回收率分别为97.66%、97.32%、97.88%、97.67%、98.35%、97.52%、98.34%、98.41%、98.03%、97.81%、98.19%,RSD值分别为0.5%、1.2%、0.8%、0.5%、0.5%、0.9%、0.3%、0.5%、0.4%、0.6%、1.0%,表明该方法准确、可靠。
2.4样品测定 精密称定4批样品粉末各2.0 g,按照2.2.2项下方法制备供试品溶液,精密吸取混合对照品溶液和供试品溶液各2 μL,按照2.1项下色谱条件测定,计算各成分含量,结果见表4。

表4 样品含量测定结果 (n=4)
3 讨论
3.1检测器的选择 HPLC法同时检测11种成分,存在前处理复杂、基线分离难、最大吸收波长不同等问题,而质谱法具有灵敏度高、准确性好、选择性强、线性范围宽的特点。此外,以多反应监测模式进行含量测定,同时扫描正负离子对,相较于紫外检测器,检验的时间更短,结果更准确。
3.2监测离子对的选择 质谱多反应监测模式以特征离子为目标,选择合适的母离子和子离子是检测的关键。本实验先以全扫描模式确定母离子,再优化碎裂能量和碰撞能量,确定信号响应最强的子离子,最终为11种成分各确定1对定量离子。
3.3流动相的选择 分别比较了乙腈-水、乙腈-1 mL·L-1甲酸、甲醇-水、甲醇-1 mL·L-1甲酸作为流动相的检测结果,发现乙腈和甲醇均能达到较好的分离效果,并且乙腈较甲醇具有更强的洗脱能力和更低的洗脱压力。少量甲酸的加入既改善了峰形,又提高了响应,因此,最终选用乙腈-1 mL·L-1甲酸作为流动相。
3.4提取溶剂的选择 由于有效成分多为酚酸和皂苷类化合物,二者在水和甲醇中溶解度高,比较甲醇、体积分数为70%的甲醇、体积分数为50%的甲醇的提取效果,发现体积分数为70%的甲醇的提取效果最好,干扰较少,最终选用体积分数为70%的甲醇作为提取溶剂。
3.5提取方法的选择 比较超声和回流2种提取方法,结果显示,在相同提取时间下,回流提取液的含量高于超声提取液,最终选用回流提取法。
3.6提取时间的选择 比较回流提取30、60、90 min的结果,发现提取60 min时,各成分含量基本一致,最终选用提取时间为60 min。
根据上述实验结果,本方法具有前处理简便、分析时间短、结果准确、检测成分多等特点,可为杞菊地黄丸的质量控制提供参考。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
中国外汇(2019年10期)2019-08-27
中国外汇(2019年10期)2019-08-27
中国外汇(2019年22期)2019-05-21
中国外汇(2019年21期)2019-05-21
中国科技纵横(2018年2期)2018-11-29
中成药(2018年7期)2018-08-04
中国洗涤用品工业(2015年8期)2015-02-28
无机化学学报(2014年9期)2014-02-28
中国医疗器械杂志(2013年5期)2013-12-05
