
耳聋基因芯片法检测在耳声发射不合格的非综合征型感音神经性耳聋新生儿筛查中的应用价值
2021-11-16 05:23:32江玉刚耿敏彦
陕西医学杂志 2021年11期
江玉刚,耿敏彦
(西安工会医院耳鼻喉科,陕西 西安 710100)
近年来,新生儿听力筛查已在我国广泛推广及应用,早期对新生儿听力做出精准的评估,可根据评估结果为其制定及时有效的治疗方案,故新生儿听力筛查具有重要的临床价值[1]。诸多新生儿听力筛查研究提示,部分新生儿出生时即被诊断为耳声发射不合格,而这部分新生儿可由单一基因突变或不同基因的复合突变导致,且与环境相关,部分新生儿由基因与环境两种因素共同造成[2-3]。已有相关研究[4]证实,50%的耳聋患儿与遗传因素相关,其中70%的遗传性耳聋以耳聋外不伴随其他临床症状即非综合征耳聋。在我国,大部分耳声发射不合格的非综合征型感音神经性耳聋新生儿的突变基因为GJB2、GJB3、SLC26A4及mtDNA12s rRNA等,而以上突变基因为耳声发射不合格的非综合征型感音神经性耳聋新生儿的基因筛查及诊断提供了理论依据。现临床常采用直接测序、限制性片段长度多态性分析、酶切、DHPLC等常规基因诊断方法,但由于上述方法存在价格昂贵、耗时费力及不能定性等局限性,且无法检测不同基因的多个突变位点。为实现临床快速检测,急需建立一种精准的基因突变检测法。而耳聋基因芯片法是一种耳聋基因筛查工具,其主要检测特点为其固有的高效平行,其高度与耳聋基因高遗传异质性的特点相契合,且具有极高的检测耳聋基因的潜力。但关于耳聋基因芯片在耳声发射不合格的非综合征型感音神经性耳聋新生儿筛查中的应用甚少。基于此,本研究将探讨耳聋基因芯片法在耳声发射不合格的非综合征型感音神经性耳聋新生儿筛查中的应用价值,以期为临床提供借鉴。
1 资料与方法
1.1 一般资料 选择2016年1月至2020年2月于我院出生的70例耳声发射不合格的非综合征型感音神经性耳聋新生儿为研究对象。其中男39例,女31例;年龄3~27 d,平均(12.83±3.21)d;体重2500~3990 g,平均(2760±30)g。病例纳入标准:①符合听力检查诊断标准:新生儿出生3 d后采用耳声发射法(Otoacousticemisson,OAE)和自动听性脑干反应法(Automatic auditory brainstem response,AABR)进行听力筛查,判定结果为“refer”或“pass”,若初次筛查未通过则需在新生儿42 d时再次复查,两者均未通过即为耳声发射不合格的非综合征型感音神经性耳聋。②临床资料完整;③均采集静脉血进行;④足月、单胎患儿;⑤本研究经医院伦理委员会的批准,且患儿家属均知情同意。排除标准:临床资料不完整;体重过低或巨大儿;早产儿。
1.2 研究方法
1.2.1 耳聋基因芯片法检测:①试剂及仪器:北京天根生化科技有限公司生产的小量基因组DNA提取试剂盒;博奥生物技术有限公司生产的基因扩增仪(BIOERTC-96/G/H)、芯片扫描仪(Lux-ScanTM10 k/B)、芯片洗干仪(Slide-WasherTM8)、芯片杂交仪(BiomixerTMⅡ)、PCR仪(Cycler96)。②DNA提取:采用含枸橼酸钠抗凝剂的真空采血管收集患儿的足跟血3滴,采用试剂盒提取DNA,其操作步骤需严格根据说明书进行操作,DNA定量和纯度采用BECKMAN DU800紫外分光光度计检测。③PCR反应:PCR反应体系为25 μl,将其A、B两个反应体系,多重PCR扩增采用BIOER TC-96/G/H。④杂交:在芯片的点样区域内加入PCR变性产物混合液,将杂交盒密闭,在水平位快速放进50 ℃预热的杂交仪中杂交1 h。⑤洗片、甩干及扫描:经SLidewasher8芯片洗干仪与按照规程配置好洗涤液Ⅰ和洗涤液Ⅱ相连接,摇床洗涤2 min。在干仓内放入芯片,置入离心机以1600 r/min离心2 min甩干。芯片采用Lux-ScanTM10 k/B微陈列芯片扫描仪,激光扫描强度设置为90和激发波长为532 nm。⑥检测SLC26A4基因、GJB2编码区序列及线粒体DNA 12SrDNA A1555G点突变:设计引物,PCR扩增及直接测序。其中SLC26A4基因序列:对突变热点区域第7+8、第19外显子进行筛查,若发现单杂合突变则需对第10、15、17及3外显子进行筛查,并对其余下外显子进行筛查,直至出现另一个突变或对其全部外显子进行筛查;若发现复合杂合突变或纯合则可停止检测。Prev-DAF药物性耳聋基因诊断试剂盒检测线粒体DNA 12SrDNA A1555G 点突变。
1.2.2 酶切或测序法:于15 μl PCR扩增产物中加入3 μl的酶切缓冲液(10×)、1 μl的Taq Ⅰ酶,补充蒸馏水到30 μl,即总体系30 μl。65 ℃水浴箱酶切过夜,酶切产物采用1%琼脂糖凝胶电泳检测。PCR产物送至诺和生物科技有限公司测序,具体步骤见该公司操作手册。
1.2.3 结果评定:DNA序列为两列(a、b),若探针检测同个位点a列出现阴性信号,b列为阳性信号,则判读该位点为突变纯合型;若探针检测同个位点a列出现阳信号,b列为阳性信号,则判读该位点为杂合突变型;若探针检测同个位点a列出现阳性信号,b列为阳性信号,则判读该位点为野生型;若在不同位点出现以上多种情况,则判读该位点为复合突变型。
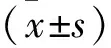
2 结 果
2.1 70例新生儿检测结果 经耳聋基因芯片检测70例患儿的阳性率为50.00%;经酶切或测序法70例患儿的阳性率为54.29%,组间比较差异无统计学意义(P>0.05)。见表1。

表1 70例新生儿检测结果 [例(%)]
2.2 不同检测方法对GJB2基因突变等基因检测结果 对176del16、35delG、299-300delAT、235delC的GJB2基因突变位点的芯片点阵,其中测序法分别检出0个、1个、59个、9个;基因芯片法分别检出等位基因突变0个、1个、57个及9个;后3个的等位基因突变检出率,测序法与基因芯片法的符合率依次为100%(1/1)、100%(9/9)及96.61%(57/59)。由于235delC点位在芯片结果中采用肉眼观察被误判为单杂合突变,故符合率为96.61%。
2.3 不同检测方法对SLC26A4等位基因突变检测结果 IVS7-2A>G、2168A>G的SLC26A4等位基因突变的芯片点阵,其中测序法分别检出等位基因突变6个和27个;基因芯片法的分别检出等位基因突变6个和27个。测序法与基因芯片法的等位基因突变的符合率为100%(6/6、27/27)。
2.4 随访结果 70例患儿均被告知于3个月龄时到门诊进行常规听力诊断及随访,其中4例mtDNAA155G及23例GJB2基因突变的患儿被称为中度耳聋,11例SLC26A4突变患儿被称为重度耳聋。其余31例耳声发射不合格的新生儿非综合征型感音神经性耳聋患儿被诊断为听力未见异常及1例IVS7-2A>G和299-300delAT双杂合突变者为正常,且后期每半年对其进行听力检测一次。见表2。
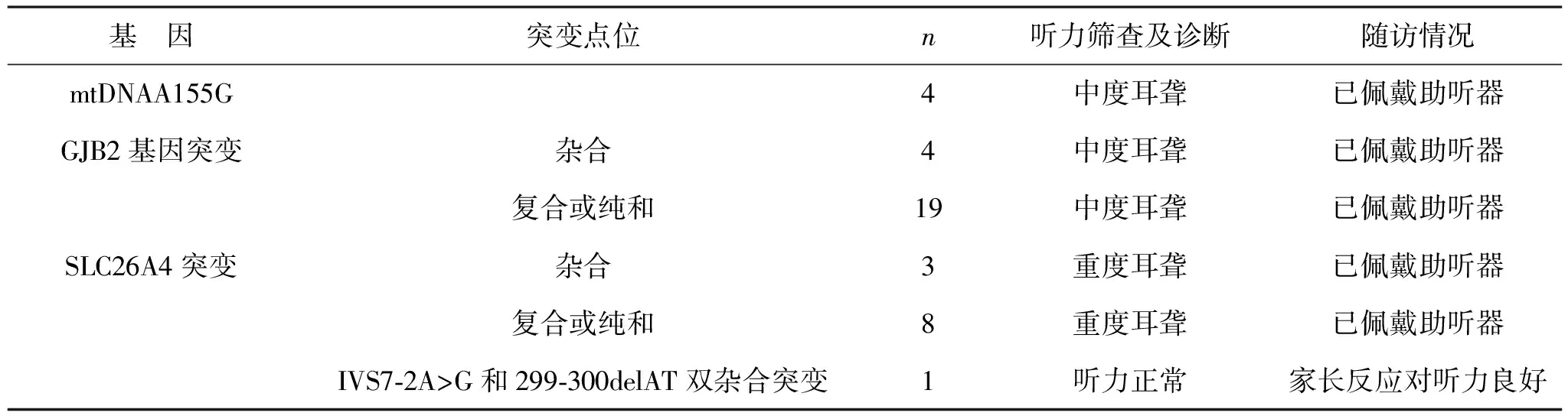
表2 70例患儿随访结果
3 讨 论
2005年据世界卫生组织估计,全球听力障碍患者可高达2.78亿。而我国约有2780万人为听力障碍患者,约占障碍群体的33.5%。其中新生儿耳声发射不合格的发生率约为1%~3%,是临床最为常见的先天缺陷之一[5]。新生儿听力筛查可及早发现听力存在不合格的患儿,进而在语言发育期给予有效干预,避免损伤语言发育[6-7]。由于耳聋的遗传异质性较高,大部分耳声发射不合格的新生儿相关基因及位点不断被发现,但常规检测方法对多突变位点不敏感。而遗传性耳聋基因诊断芯片针对迟发性耳聋、药物性耳聋等4个基因中的9个突变位点具有极高的敏感性,其主要特点为操作简单、自动化及通量高等[8]。故本研究将探讨耳聋基因芯片在耳声发射不合格的新生儿非综合征型感音神经性耳聋患儿中的应用价值,从基因水平对耳声发射不合格的新生儿非综合征型感音神经性耳聋患儿的发病原因进行明确,以便为其提供及时的治疗及指导。
本研究结果显示,经耳聋基因芯片法检测70例耳声发射不合格的非综合征型感音神经性耳聋新生儿的阳性率为50.00%,经酶切或测序法检测70的阳性率为54.29%,两者比较差异无统计学意义,但单杂合突变的患者提示可能为遗传性耳聋,可建议进一步进行耳聋基因筛查。与前人研究结果相似[9-10]。同时在本研究结果中,未检测出707T>C和167delT突变,间接提示了耳声发射不合格的新生儿非综合征型感音神经性耳聋患儿的种族异质性。GJB2基因编码连接蛋白常表达于人体内的耳蜗及皮肤组织内,该基因突变后K+进入机体淋巴液内致使淋巴液循环受到影响,引发感音神经性耳聋[11-12]。SLC26A4基因突变会引发Pendred和DFNB4综合征常染色体隐性耳聋[13-15]。而非综合性常染色体隐性遗传耳聋中最为常见的致病基因为上述两种基因突变[16-18]。本研究结果显示,对176del16、35delG、299-300delAT、235delC的GJB2基因突变位点的芯片点阵,酶切或测序法与基因芯片法的符合率依次为100%、100%及96.61%。由于235delC点位在芯片结果中采用肉眼观察易被误判为单杂合突变。IVS7-2A>G、2168A>G的SLC26A4等位基因突变的芯片点阵,酶切或测序法与基因芯片法的等位基因突变的符合率为100%,说明液相酶促反应具有极高的特异性,在耳聋基因芯片法进行读片时出现肉眼误判,这与芯片标签(Tag)探针与带荧光PCR产物的非特异性杂交导致增强信号,故被判为阳性,对此点阵的Tag序列进行优换,可避免降低出现同样的错误[19]。并且统计本次研究中芯片检测结果中的突变型及野生型的平均荧光信号值,对两者间的CUT-OFF进行设定,并采用自动判读软件,可避免出现肉眼观察出现的错误,且可更直观、快捷。基因芯片法每张芯片可同时检测多个个体,且每次能同时检测多张芯片,对仪器要求不高、试剂价格低廉,相比传统方法耗时短、通量高、成本低,能更有效的满足临床需求。
本次研究中随访结果显示,4例mtDNAA155G及23例GJB2基因突变的患儿被称为中度耳聋,11例SLC26A4突变患儿被称为重度耳聋。对12例SLC26A4突变患儿的父母进行检测其父母均为新生儿相应突变基因位点的杂合携带者,为避免再次生出耳聋患者,建议再次怀孕时应进行产前耳聋常见基因检测。由于同一基因的杂合或纯和突变会出现叠加效应,故4例mtDNAA155G及23例GJB2基因突变患儿的听力也会受到一定程度的影响。如今,经本次研究的38例耳声发射不合格的新生儿非综合征型感音神经性耳聋患儿均已佩戴助听器,听力效果良好。国内学者蔡超婵等[20]研究提示,采用人工耳蜗植入术治疗GJB2基因突变的非综合性感音神经性耳聋患儿,术后效果理想,在对其进行制定最佳方案时可采用基因诊断。另外经笔者总结,针对GJB2基因突变的患儿在成长过程中需注意保护患儿头部,防止患儿头部受伤、远离噪音、用力咳嗽、憋气及擤鼻,若发现听力发生异常后需及时就医。其余31例耳声发射不合格的新生儿非综合征型感音神经性耳聋患儿被诊断为听力未见异常及1例IVS7-2A>G和299-300delAT双杂合突变患儿,且后期每半年对其进行听力检测一次。这可能是由于该部分新生儿外耳道有羊水或胎脂致使听力筛查时耳声发射不合格造成结果为“不合格”假阳性,但避免出现漏检,故针对该部分患儿每隔半年再次进行听力筛查,以达到早发现、早治疗,并对其进行随访至成年。
总而言之,相比酶切或测序法,耳聋基因芯片法成本低、准确性高、操作简单快速且更具标准化,能满足临床对常见耳聋基因检测需求,值得临床推广使用。
猜你喜欢
现代实用医学(2022年2期)2022-04-02 03:19:12
开卷有益·求医问药(2021年12期)2021-12-29 00:34:55
中医眼耳鼻喉杂志(2021年2期)2021-07-21 08:53:28
今日农业(2021年4期)2021-06-09 06:59:56
中国医疗器械信息(2021年16期)2021-04-02 08:19:01
山西大同大学学报(自然科学版)(2020年2期)2020-12-09 01:15:42
快乐语文(2018年31期)2018-03-01 11:22:56
现代检验医学杂志(2016年4期)2016-11-15 02:01:00
中国药物经济学(2015年9期)2015-12-08 03:32:56
哈尔滨医药(2015年3期)2015-12-01 03:57:44
