
KCNB1基因新发杂合错义突变致癫痫性脑病临床诊治
2021-11-15 07:58金爱琴
中国临床医学 2021年5期
李 斌, 金爱琴, 周 辉
南通大学附属医院小儿内科,南通 226600
发育和癫痫性脑病(developmental and epileptic encephalopathy, DEE)是指因发育性脑损伤伴频繁痫性活动引起的智力倒退和后续发育迟缓。DEE作为一种神经发育性疾病,在儿童中较多见。癫痫在智力障碍人群中发生率较高,为20%~30%,且癫痫发生率与患者的智力障碍程度密切相关[1-2]。约16%的癫痫患儿有程度不等的智力损伤[3]。DEE患者临床表现主要有不同程度的社交行为障碍、认知障碍以及运动和语言障碍。智力障碍主要为癫痫相关活动(癫痫发作和脑电图异常)所致的脑发育异常[4]。
神经系统兴奋性的损伤和调控与离子通道有关,其中DEE的发生可能与离子通道编码异常有关[5],而离子通道的异常与基因突变有关[6]。有研究[7]发现,钾电压门控通道亚家族B成员1(KCNB1)基因发生突变,可引起KCNB1编码的电压依赖性钾通道Kv2.1发生异常,从而导致DEE的发生。本院近来收治1例KCNB1基因突变的DEE患儿,现报告其诊治经过,同时复习相关文献,以期提高临床对精神运动发育迟缓合并癫痫的认识。
1 资料与方法
1.1 既往病史 患儿,女性,3岁7月龄,因“反复惊厥发作伴精神运动发育迟缓”于2019年9月南通大学附属医院门诊就诊。患儿23月龄时曾因“发热2 d,惊厥30 min”于2018年2月某妇幼保健院住院。惊厥发作时表现为两眼上翻,四肢抽搐用力、口周发绀、意识不清,发作持续30 min;病程中有流涕,常规脑电图显示无异常,头颅MRI未见异常。诊断为上呼吸道感染,热性惊厥,癫痫持续状态,白细胞减少症。患儿住院3 d后出院,出院时医师建议长程脑电图,家长未重视;出院后未予以抗癫痫治疗,随后17个月抽搐未发作。患儿3岁6月龄时因“无热惊厥1次”于2019年8月再次于该妇幼保健院住院,惊厥发作时表现为两眼凝视、四肢抽搐用力、口周发绀、意识不清,发作持续约1 min。常规脑电图:大脑皮质中央区少量尖波、尖慢波发放;头颅MRI未见异常。住院5 d好转出院,未予以抗癫痫治疗。2019年9月再次出现无热惊厥,于南通大学附属医院进一步检查。
1.2 诊治经过
1.2.1 常规诊治经过 患儿母亲G1P1,足月剖宫产,否认产伤窒息史,孕期身体欠佳(保胎),无癫痫家族史。患儿生长发育落后,5月龄竖头,10月龄独坐,14月龄独站,18月龄独走;18月龄说话叫人,言语发育缓慢,与人沟通少,眼神交流少,不喜欢与同龄儿童玩耍。查体:神志清,精神可,无特殊面容,皮肤无异常,心肺腹无异常,肌张力低,肌力5级,腱反射可引出,病理征(-)。实验室检查:血常规、肝肾功能、电解质、血糖正常。心电图无异常。遗传代谢病筛查:血、尿串联质谱无异常。生长发育落后:儿童智能发育筛查测验(DST)量表中,发育商(DQ)<49、智力商数(MI)<49。常规脑电地形图:在10%水合氯醛灌肠诱导睡眠状态下描记,各区均为2~5 Hz、约150 μV,负荷多量低幅快波,伴睡眠纺锤波和顶尖波;两半球大致对称。病理波:各区见多量同步及非同步性3~4 Hz高波幅尖慢、棘慢、多棘慢波单发、簇发,前头部著。脑电地形图:各区δ、θ频带功率增高。2019年9月30日视频脑电图(VEEG)示双侧额、中央、顶、颞、中央中线区尖慢波、棘慢波发放(图1A)。诊断:癫痫合并DEE;建议抗癫痫治疗及康复训练。抗癫痫治疗:左乙拉西坦(LEV)1.5 mL每日2次(LEV 21 mg·kg-1·d-1)。患儿癫痫半年未发作。2020年4月22日,患儿在患急性上呼吸道感染后又发作1次,时间不长,发作形式改变,表现为眼斜视发呆、喉中有痰声、恶心想呕吐、意识模糊、口周发绀,无四肢抽搐,持续约30 s,予LEV逐渐加量至3.0 mL每日2次(LEV 42 mg·kg-1·d-1),治疗后仍反复发作,1~2次/周。2020年4月29日复查VEEG(图1B),示双侧前额、额、中央、顶、颞区尖慢波、棘慢波发放。
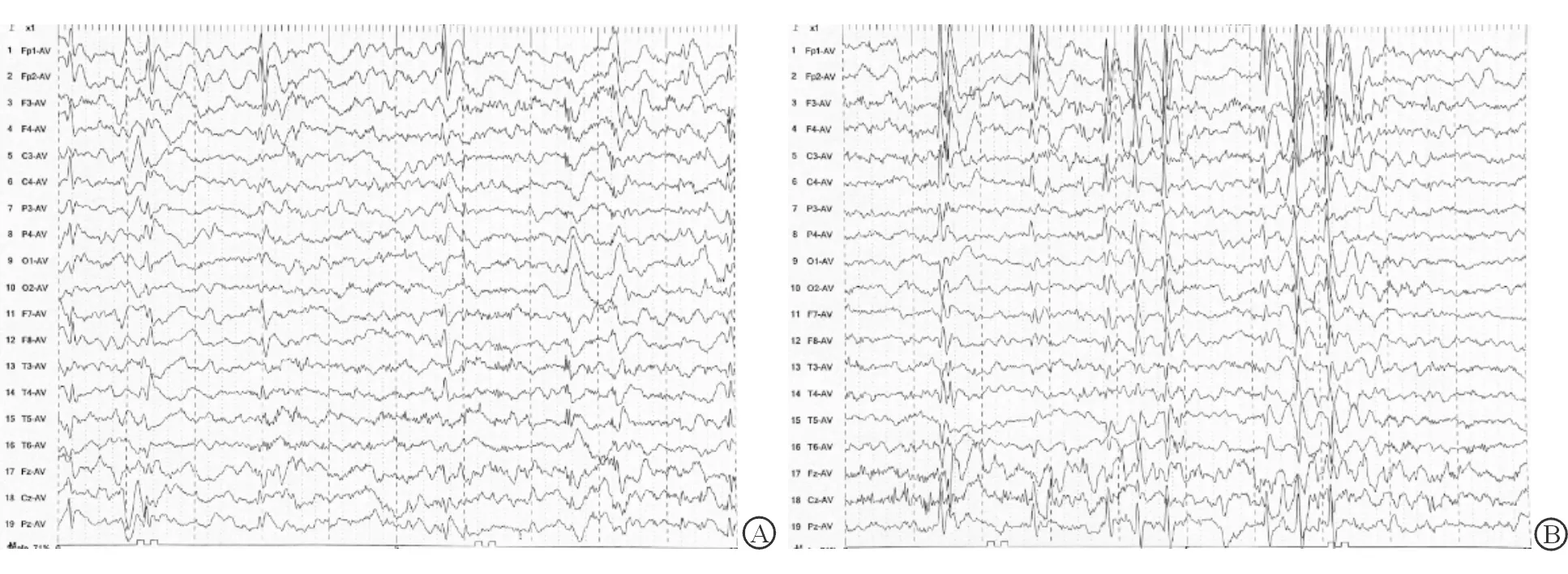
图1 患儿治疗前后VEEG
1.2.2 基因检测 为进一步明确病因,经患儿家属知情同意及南通大学附属医院医学伦理委员会审核批准(202005004)后,委托明码(上海)生物科技有限公司行外周血基因Sanger测序。检测结果(图2):KCNB1基因新生杂合错义突变,基因位于20号染色体,突变位点为c.904A>G(p.M302V) ,即该位点A(腺嘌呤)突变为G(鸟瞟呤),使M(甲硫氨酸)突变为V(缬氨酸)。KCNB1基因是癫痫性脑病26型(早期婴儿型癫痫性脑病,EIEE)的致病基因。EIEE呈常染色体显性遗传,主要表现为顽固性癫痫发作、神经发育障碍,预后不良。本例患儿的临床表现与该突变所致表现相符。Sanger测序未发现患儿父母基因组该位点存在上述变异,提示该突变为患儿新发。
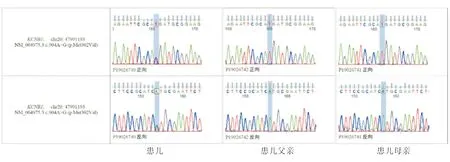
图2 患儿及其父、母亲基因组Sanger测序图
2 讨 论
癫痫按病因发生可分为遗传性癫痫和继发性癫痫,在儿童癫痫中以遗传性癫痫较为常见。与遗传性癫痫发生相关的基因主要有2类:离子通道相关基因和非离子通道相关基因。其中编码钾离子通道的基因在癫痫易感基因中比例较高[8]。钾通道相关的神经系统疾病通常与癫痫有关[9]。在编码钾通道成分的基因中,KCNB1编码电压门控钾通道Kv2.1的α亚基,该亚基由858个氨基酸组成,包含6个跨膜片段(S1~6),位于质膜上。Kv2.1是Kv2复合体的一个亚单元,包括Kv2.1和Kv2.2。Kv通道的基本结构包括选择性滤波器、电压传感和门控元件,大部分是膜内元件。KCNB1在中枢神经系统中表达,尤其在新皮层和锥体神经元树突中表达丰富。KCNB1缺失小鼠模型神经元过度兴奋,且Kv2.1在其海马神经元网络稳态调节中起关键作用[10]。此外,KCNB1基因编码Kv2.1的孔隙域和电压感应β亚单位,该亚单位在整个大脑中表达,并在调节神经元兴奋性、促进动作电位复极和神经元活动的动态调节中发挥重要作用[11]。Kv2.1/KCNB1在大脑的兴奋性和抑制性神经元中均有表达,并且在海马和皮质锥体神经元中主要作用为延迟整流钾离子电流的发生[12]。
Kv2.1可以通过磷酸化调节神经元活动,进而调控平衡神经元兴奋性。KCNB1基因突变常引起神经系统性疾病发生,其突变位点较为广泛,绝大部分为错义突变,而且为功能丧失(LOF)突变[13],且发生错义变异的癫痫患者预后更差[14]。KCNB1基因突变可引起Kv2.1功能降低,且Kv2.1功能下降程度与患者临床表现的严重程度正相关。有研究[13]对新杂合子KCNB1突变脑病和神经发育障碍患者进行分析发现,错义突变可能引起静息膜电位去极化和膜复极受损,进而提高神经元兴奋性。然而,KCNB1基因突变对神经元活动的影响受其他离子通道和分子性质等的影响[15],癫痫患者KCNB1突变频率仍有待阐明。
遗传学研究[11]表明,Kv基因突变(包括KNCQ2、KCNQ3、KCNA2、KCNA1、KCNC1、KCNH1、KCNMA1、HCN2和KCNT1)多与癫痫病相关,导致的癫痫病严重程度从良性到EIEE不等。OMIM将 KCNB1突变引起的严重癫痫疾病称为EIEE 26型。EIEE 26型呈常染色体显性遗传,一般在1岁内发病,临床表现为肌张力低下,认知发育迟缓,但认知能力可随年龄的增长提高[16]。KCNB1相关表现包括婴儿全身性或局灶性的癫痫发作,可导致EIEE,包括West、Lennox-Gastaut和Jeavons综合征。回顾性分析表明,癫痫发作之前,患儿认知和运动功能常延迟,并发展成严重的、持续的智力损伤。因此,KCNB1突变与弥漫性脑功能不全相关,包括癫痫发作、运动和认知障碍[11]。KCNB1脑病表型不同,如DEE、不伴癫痫的发育性脑病[15]。KCNB1脑病表型可能与KCNB1离子通道结构域的新生错义变体以及该结构域或C端功能缺失变体有关,但蛋白质变异类型和位置不完全与疾病的严重程度相关[17]。DEE是严重癫痫的一种,特征为儿童期早发难治性癫痫和发育停滞。EIEE是DEE的最早形式之一,婴儿早期表现为癫痫、痉挛频繁发作。KCNB1脑病以DEE患者的认知损害更严重,而KCNB1脑病患者总体远期预后多较差。KCNB1脑病患儿头颅MRI常正常,EEG可出现基本正常背景活动上的慢性、广泛性或多灶性的癫痫波[18],但均无特异性。
本例患儿KCNB1突变是一种新生杂合错义突变,突变位点为c.904A>G(p.M302V) 。在人类基因组突变数据库(HGMD)、在线人类孟德尔遗传数据库(OMIM)、dbSNP及GenomeAD等数据库检索,未发现该位点的相关报道,提示不属于多态性变化,此次为首次报告,扩充了KCNB1脑病的基因突变谱。应用SIFT、Polyphen 2 2种软件进行预测,结果提示该变异可改变蛋白质特性,从而影响蛋白质功能,为致病性有害变异。本例患儿的临床表现与KCNB1突变导致的肌张力低下、认知发育迟缓的表现符合,而Sanger测序未发现父母携带上述变异,提示为患儿的新发变异,推测该突变位于电压传感和孔隙域。膜片钳技术检测显示,Neuro2a细胞和原代皮层神经元上的这种Kv2.1突变体导致的功能异常与以往报道[11]的Kv2.1突变不同,能抑制产生尖峰间的高幅度电压,从而不同程度得抑制皮层神经元的重复动作电位(AP)放电。
综上所述,KCNB1脑病是KCNB1基因突变导致的常染色体显性遗传病,患者存在严重的认知损害,远期预后较差。对于不明原因智力倒退、运动发育迟滞合并难治性癫痫发作的患儿,应尽早行遗传学检测,以对患儿病情进行准确评估,为精准治疗提供依据。本例报告扩充了KCNB1脑病的基因突变谱。KCNB1脑病基因突变谱的不断完善可为产前诊断及遗传咨询提供更多内容。
利益冲突:所有作者声明不存在利益冲突。
猜你喜欢
现代农业科技(2022年22期)2022-12-01
——人工离子通道
大学化学(2022年9期)2022-10-20
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国典型病例大全(2022年10期)2022-05-10
保健文汇(2021年5期)2021-05-22
陕西医学杂志(2020年10期)2020-12-27
科学之谜(2018年9期)2018-12-17
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
中国医学创新(2016年33期)2017-02-28
