
高效液相色谱法检测猪肉中4 种氟喹诺酮类药物残留方法的优化
2021-11-13 07:17田雨超谷学佳
现代畜牧兽医 2021年10期
田雨超,谷学佳,薛 丽
(1.北京市昌平区农产品监测检测中心,北京 102200;2.北京市大兴区农业农村局动物疾病控制中心,北京 102600)
喹诺酮类药物(4-quinolones)是人工合成抗菌药,以抑制细菌的DNA回旋酶为机理,抗菌作用强大,抗菌谱较广。一般将这类药物分为4代,从第3代开始加入氟原子,称为氟喹诺酮类。至今已经有20个氟喹诺酮类药物品种上市,大多用于畜禽和水产养殖行业。氟喹诺酮类药物在兽医临床上最常用的有诺氟沙星、环丙沙星、恩诺沙星、氧氟沙星、丹诺沙星、马波沙星等[1]。氟喹诺酮类药物常见的不良反应为幼畜软骨变性,大量使用会产生结晶损伤尿道及胃肠道反应。这类药物的不正确使用和不遵循休药期会导致药物残留,也会使致病菌产生耐药性,间接危害人类健康[2]。该药物长期大量累积会造成肝损害,危及生命。多个国家和地区均规定了氟喹诺酮类药物的最高残留限量[3]。国外对动物食品中喹诺酮类药物残留的研究主要集中在前处理方法以及新材料的应用,或者开发对多种类药物残留的一次性综合检验新方法,如微生物法的应用等[4-7];而国内研究则集中在样品前处理方法的条件优化上,一般标准方法的操作步骤及色谱条件[8],很多细节甚至关键步骤需要操作者在实际应用中去摸索。标准方法经改进细化,不仅成本较低、前处理简单易行,而且给基层操作者带来方便和可应用性参考。根据基层检测单位的实际情况,为节省成本和提高工作效率,优化色谱条件和简化前处理步骤成为首选改进因素。目前,在动物产品的检测中,很多推荐使用液相色谱-串联质谱法。但由于基层单位仪器设备条件有限,《动物性食品中氟喹诺酮类药物残留检测高效液相色谱法》(农业部1025 号公告-14-2008)[9]成为基层单位主要检测方法。本试验以此标准为基础,简单细化了试验前处理步骤,优化了检测过程,减少所需耗材较大幅度降低成本,尤其对大批量样品检测,节省了处理时间。本方法的灵敏度和回收率均可满足我国现行兽药残留检测分析的要求。
1 材料与方法
1.1 试验材料
试验使用的猪肉购自二商大红门五肉联,经检测,样品均不含诺氟沙星、环丙沙星和恩诺沙星、沙拉沙星药物残留。
1.2 仪器与试剂
高效液相色谱仪1260(荧光检测器)(安捷伦)、B-400组织捣碎机(瑞士步琦)、GT10-1高速台式离心机(北京时代北利离心机有限公司)、CAX-371 高速冷冻离心机(TOMX)、ME104E/02 天平(Mettler Toled)、TTL-DCII 氮吹仪、MX-F 漩涡振荡器(Scilogex)、LPD2500 多管混合仪(莱普特科学仪器(北京有限公司))、Eppendorf 微量移液器(Thermo Finnpiptte)、KS康氏振荡器;PHS-3C pH计(雷滋)、ME55 型电子天平(瑞士梅特勒-托利多仪器有限公司)、超声波仪、Waters固相萃取柱HLB(3 mL)。
乙腈、甲醇、异丙醇(色谱纯,Fisher Chemical);磷酸(分析纯,国药集团化学试剂有限公司);三乙胺(分析纯,国药集团化学试剂有限公司);磷酸二氢钾(分析纯,国药集团化学试剂有限公司);氯化钾(分析纯,国药集团化学试剂有限公司);氢氧化钠(分析纯,北京化工厂);超纯水;蒸馏水(屈臣氏)。
标准品:达氟沙星标准品,纯度94.27%;恩诺沙星标准品,纯度99.00%;环丙沙星标准品,纯度92.31%;沙拉沙星标准品,纯度85.82%,均购自Dr.Ehrenstorfer GmbH。
1.3 试液配制
环丙沙星、恩诺沙星是光敏性药物,应避光保存。配制过程中应在暗处进行,防止药物光解,配制完后储存在棕色避光瓶中。
分别称取对照品恩诺沙星10.00 mg、环丙沙星11.10 mg、沙拉沙星10.95 mg,置于同一个10 mL 容量瓶中,使用0.03 mol/L 的氢氧化钠溶液溶解定容至刻度,制得质量浓度均为1 g/L 的混合储备溶液。准确称取达氟沙星对照品达氟沙星12.69 mg,0.03 mol/L 的氢氧化钠溶液溶解定容至10 mL,使其质量浓度为1 g/L,置2~8 ℃冰箱中保存,备用,有效期3个月。取上述恩诺沙星、环丙沙星和沙拉沙星的混合储备液1.00 mL,达氟沙星储备液0.20 mL,置于同一个10 mL 容量瓶中,用乙腈定容至刻度,配置成恩诺沙星、环丙沙星、沙拉沙星的质量浓度为100 mg/L,达氟沙星质量浓度为20 mg/L 的混合标准溶液。取上述混合储备液1.00 mL,置于1 个10 mL 容量瓶中,使用乙腈定容至刻度,配置成恩诺沙星、环丙沙星、沙拉沙星的质量浓度为10 µg/mL,达氟沙星质量浓度为2 mg/L 的混合标准溶液。使用流动相配制成所需要浓度的混合标准溶液以备使用,标液配制稀释级不要太大,标准曲线浓度尽量不要从上一浓度梯度稀释,避免标液配制出现偏差。
5.0mol/L 氢氧化钠溶液:取氢氧化钠饱和液28 mL,加水稀释至100 mL,混匀(根据需要适量配制)。
0.03mol/L 氢氧化钠溶液:取5.0 mol/L 氢氧化钠液0.6 mL,加水稀释至100 mL,混匀。
磷酸/三乙胺溶液:取浓磷酸3.4 mL,加水稀释1 000 mL,使用三乙胺调pH 值至2.4。现用现配(易产生沉淀)。
磷酸盐缓冲溶液(用于肌肉、脂肪组织):取磷酸二氢钾6.8 g,加水溶解并稀释至500 mL,使用5.0 mol/L氢氧化钠溶液调节pH值至7.0。
洗脱液:取乙腈20 mL,使用0.05 mol/L磷酸/三乙胺溶液稀释至100 mL,混匀。取乙腈18 mL,使用0.05 mol/L磷酸/三乙胺溶液稀释至100 mL混匀,备用。
1.4 样品处理
称取(2.00±0.02)g 猪肉样品,加10.0 mL 磷酸盐缓冲液(pH 值=7),涡旋混匀,中速振荡5 min,10 000 r/min 离心,取上清液到新离心管待用,再向原离心管加磷酸盐缓冲液10.0 mL,涡旋混匀,中速振荡5 min,10 000 r/min 离心,2次上清液合混匀,使用滤纸过滤,备用。
标记好的固相萃取柱先依次用甲醇、磷酸盐缓冲液各2 mL预洗。取上清液5 mL过柱,使用1 mL水淋洗,挤干。用流动相1 mL洗脱,挤干,收集洗脱液。经过滤膜过滤后作为试样溶液,供高效液相色谱法测定。
1.5 色谱条件
流动相A为0.05 mol/mL磷酸溶液/三乙胺,D为乙腈,使用前过滤脱气;流速0.8 mL/min,柱温:室温,进样量:20 µL;荧光检测器,激发波长(Kex)248 nm,发射波长(Kem)450 nm。色谱柱:ZORBAX Eclipse XDB-C18 柱(250 mm×4.6 mm,5µm)。
2 结果与分析
2.1 样品前处理条件的优化
2.1.1 样品制备环节
一般基层检测样品量大,普通的样品粉碎机费时费力,且得到均质效果不理想的样品,只能通过匀浆机进一步均质。在通过实践和同其他机构借鉴经验,本试验使用的瑞士步琦组织捣碎机B-400,30 s可以制备好样品,样品颗粒可达0.3 mm,甚至可以大块冷冻样品处理。
检测中可以直接称取上述制备样品,节省了匀浆步骤。本方法在样品制备过程中已经达到匀浆机效果,还避免2次匀浆造成药物损失,减少匀浆操作步骤时间。
2.1.2 组织提取液净化
动物源食品中基质干扰复杂,富含大量脂肪和蛋白质,因此对猪肉样品选择要求瘦肉不含有筋膜样品,但实际检测中有时无法得到理想样品。按照农业农村部的方法,样品组织提取液一般不干净呈现浑浊状态,直接加到固相萃取柱,会堵塞小柱,大大增加净化时间,并且造成药物回收率低、不稳定,导致准确率降低、残留检测难度增大。离心时采用高速冷冻离心机,低温可使脂肪凝固,还能减少药物因为离心过程中温度过高带来的损失。一般使用乙腈饱和的正己烷溶液去除脂肪等干扰物质,效果虽然较好,但是多了一步有机试剂,费时、费力、成本提高。本试验采用上述方法进行上清液用滤纸过滤,可以过掉大部分残渣,用滤纸过滤需要过柱体积的上清混合液回收率较高,不影响检测结果的准确性。经过滤后4 种农药回收率提高7.7%~13.9%,提高了过柱速度。
本试验选用固相萃取,填充剂基质是聚合物,作用和效果同C18(填充剂基质是硅胶)的固相萃取小柱,具有高吸附容量,实际工作中活化和淋洗液体积可适当调整并不影响回收率。
2.1.3 试剂配制和材料准备(见表1)
试验过程中,从标准品、提取液和净化液配制到流动配制相都需要操作者自己配制,每个细微环节都会直接影响试验结果的准确性。标液配制过程中根据《中国药品检验标准操作规范》及GB/T 601—2002《化学试剂标准滴定溶液的制备》确定选用合适量程的天平,称取量要根据化学式及纯度进行换算以去除盐酸根。标准中只有在标准曲线配制时提到了用流动相配制,根据经验建议小于1 mg/L 均用流动相配制,否则峰型不好,尤其对环丙沙星影响明显会使峰分叉。
配制时溶液用水的不同,也会影响药物提取回收率。由表1可知,本试验比较一级水和蒸馏水,一级水配制磷酸盐提取液回收率提高了3.3%~11.0%。
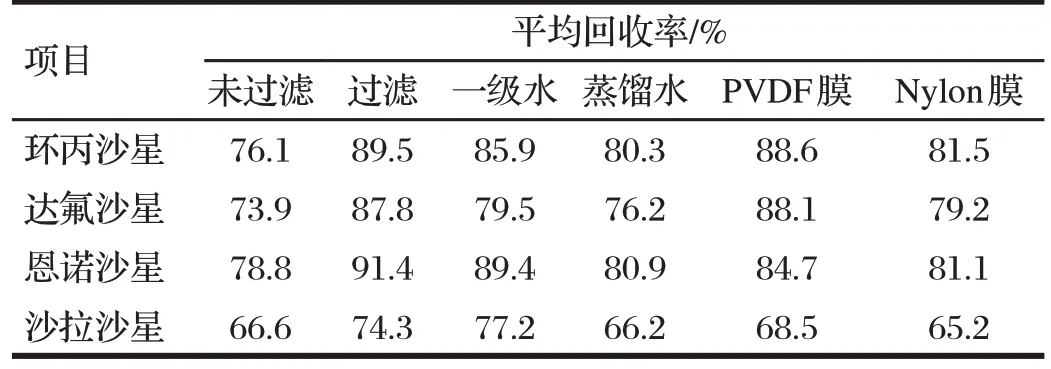
表1 样品前处理条件优化Tab.1 Optimization of sample preparation conditions
由于色谱系统的精密性和对结果分析的准确性要求,对样品的过滤显得尤为重要,过滤可以保护色谱柱系统和色谱柱,延长柱子的使用寿命,改善数据的精确度。过滤可以清除由于摩擦长生颗粒而引起的压力波动和无规律的杂质引起的极限波动,可以消除由于存在气泡对检测系统的干扰。本试验比较尼龙膜和聚偏氟乙烯膜(PVDF)。PVDF 膜使药物提取效率提高了3.3%~8.9%,尤其对达氟沙星明显提高。
2.2 色谱条件的选择(见图1、图2)
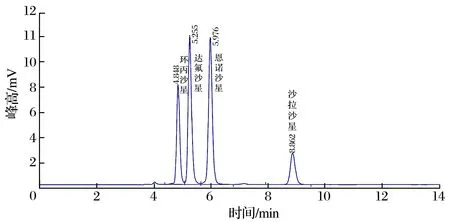
图1 流动相20∶80色谱图Fig.1 Chromatogram of mobile phase 20∶80
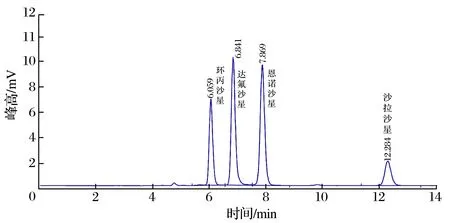
图2 流动相18∶82色谱图Fig.2 Chromatogram of mobile phase 18∶82
由图1、图2 可知,流动相中乙腈的比例越高,4 种目标物保留时间越短。考虑到干扰杂质的出峰时间和各峰的分离度,选择乙腈和水相的体积比为20∶80(标准流动相乙腈和水相的比例18∶82)。0.1 mg/L 的混合标准溶液测得环丙沙星的保留时间为4.848 min,达氟沙星为5.255 min,恩诺沙星为5.976 min,沙拉沙星为8.862 min,峰形尖锐,分离完全,无干扰峰出现。在相同色谱条件下,本试验得到的保留时间比标准方法提前3.4 min。此条件下,样品标液添加回收也得到同样色谱峰,没有样品基质干扰。
2.3 方法的线性范围与检出限(见表2)

表2 方法的线性范围与检出限Tab.2 Linear range and detection limit of the method
由表2 可知,环丙沙星、恩诺沙星、沙拉沙星在5~500 μg/L浓度范围内,达氟沙星在1~100 μg/L线性关系良好。本试验采用通过大量分析样品空白样品,独立测试次数大于10 次,计算检测结果的标准偏差(S),方法检出限结果表示为:样品空白值+4.65 S。
2.4 方法的回收率与精密度(见表3)
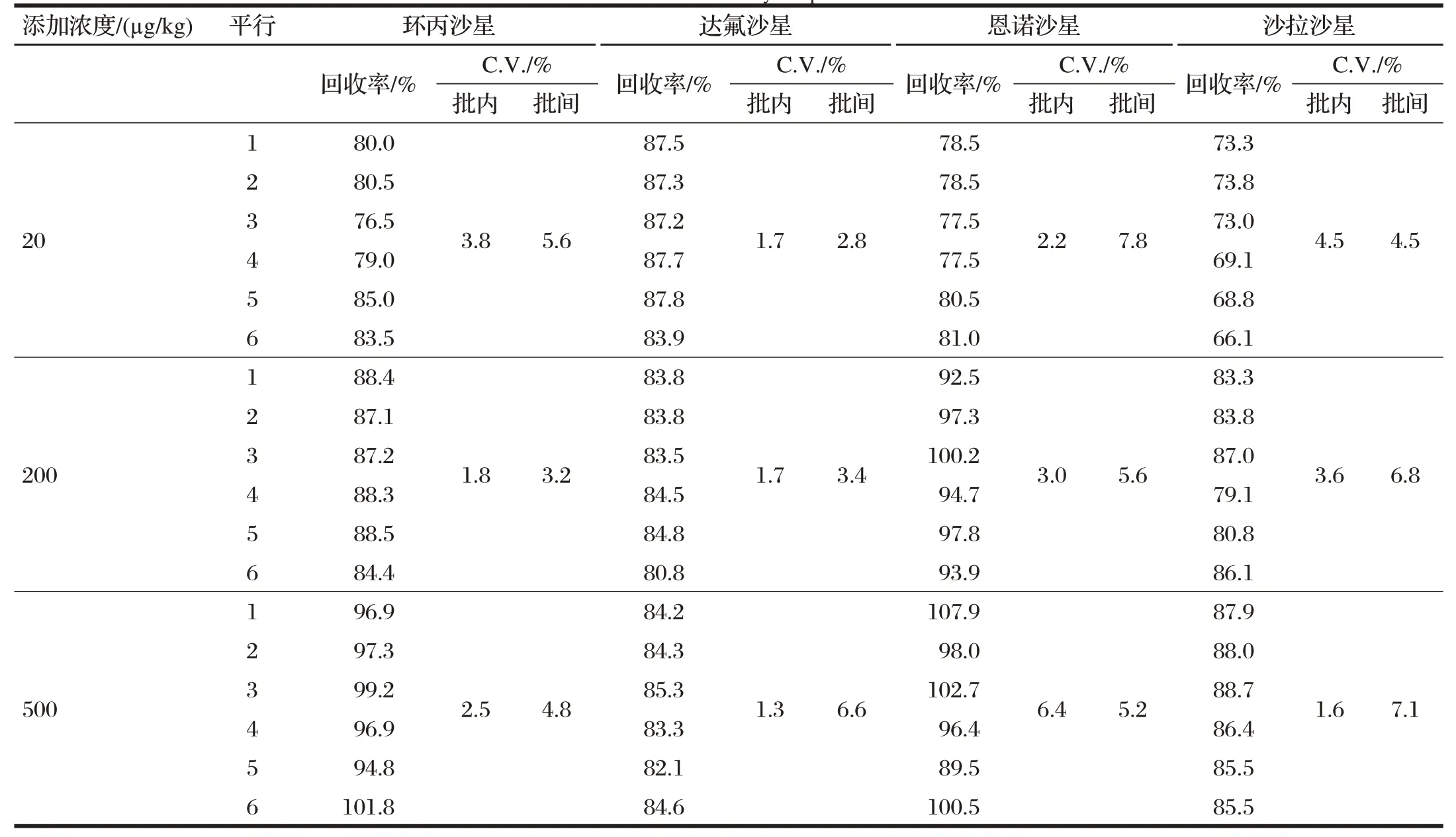
表3 方法的回收率与精密度Tab.3 Method recovery and precision
空白猪肉待测样品中分别添加3个浓度水平的混合标准液,各6个平行。由表3可知,环丙沙星3个浓度回收率在76.5%~101.8%,变异系数在1.8%~3.8%,批间变异系数为3.2%~5.6%;达氟沙星回收率在80.8%~87.8%,变异系数在1.3%~1.7%,批间变异系数为2.8%~6.6%;恩诺沙星回收率在77.5%~107.9%,变异系数在2.2%~6.4%,批间变异系数为5.2%~7.8%;沙拉沙星回收率在66.1%~88.7%,变异系数在1.6%~4.5%,批间变异系数为4.5%~7.1%。回收率及批内变异系数均在方法要求内。
3 结论
目前,大多检测方法包含样品制备、提取、净化和浓缩。在提取同时把与蛋白质结合的药物解吸附成游离药物,净化时除去样品中的干扰杂质,浓缩时达到仪器可检测的浓度范围。本试验在原有方法基础上根据实验室现有条件在各个步骤中寻找可以改进优化的点。本试验选择猪肉组织,基本组成为水分、蛋白质、脂质、碳水化合物、矿物质、维生素和酶类等,相对鸡肉、牛肉及猪肝等样品杂质干扰较少。不同组织的功用不同,在结构特点上存在差异,这些变化可能影响某些药物的回收率和干扰的检测选择并优化样品前处理方法对结果的准确性尤为重要。
高效液相色谱法在喹诺酮类药物残留的检测中被广泛应用,农业部1025 号公告-14-2008 也是各检测单位常用的方法。本试验以此标准为基础,简化了试验前处理步骤,优化了检测过程。经多次主要证明改进后优化后的方法成本降低、通用性强、方法灵敏度和回收率均可满足我国现行兽药残留检测分析的要求,可为基层检测单位提供参考。
猜你喜欢
湘潭大学自然科学学报(2022年2期)2022-07-28
北京大学学报(自然科学版)(2021年3期)2021-07-16
世界最新医学信息文摘(2021年12期)2021-06-09
环境保护与循环经济(2017年10期)2017-03-16
现代检验医学杂志(2016年2期)2016-11-14
西安理工大学学报(2016年3期)2016-11-10
国外医药(抗生素分册)(2016年5期)2016-07-12
国外医药(抗生素分册)(2016年5期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
中国卫生标准管理(2015年25期)2016-01-14
