
T细胞受体信号转导机制
2021-11-13 16:27徐晓敏李华
药学进展 2021年8期
徐晓敏 ,李华
(1. 中国科学院分子细胞科学卓越创新中心,上海 200031;2. 国科大杭州高等研究院,浙江 杭州 310000)
目前,以T细胞受体工程化T细胞(T cell receptor-engineered T cell,TCR-T)和嵌合抗原受体T细 胞(chimeric antigen receptor T cell,CAR-T)疗法为主流的过继性T细胞免疫疗法成为肿瘤免疫治疗的一大热点。通过向天然的T细胞中转入识别肿瘤抗原的T细胞受体(T cell receptor,TCR)α/β异二聚体或嵌合抗原受体(chimeric antigen receptor,CAR),T细胞便能够高效地识别靶抗原,在体内发挥抗肿瘤免疫功能。CAR-T疗法是过继性免疫疗法中研究较为火热的一员,目前已上市的产品以及处于临床阶段的CAR-T产品多针对于血液瘤,对于实体瘤的治疗效果较差。分析其中原因,主要在于实体瘤的细胞疗法存在不少难点,例如不同类型实体瘤的异质性大、缺乏独特的肿瘤相关抗原作为治疗靶点、T细胞无法定向迁移到肿瘤部位、CAR-T的增殖能力和抗凋亡能力等持续性不够以及肿瘤内复杂的微环境对TCR有抑制作用等。虽然CAR-T与TCR-T都与通用的细胞治疗兼容,并且能够诱导有效的抗肿瘤反应,但TCR-T显示出一些独特的特性,使其在治疗具有挑战性的实体瘤时具有独特的优势,但这种优势背后具体的分子机制尚不完全清楚。因此本文主要针对现有的文献报道,从TCR和CAR本身结构和信号转导差异等方面出发来理解这种临床治疗上的差异,这对于理解TCR-T针对实体瘤的治疗优势具有重要意义。
1 T细胞受体的结构
TCR是所有T细胞表面表达的由基因重排产生的天然抗原受体,它与CAR之间存在许多不同。与CAR的单链结构不同,TCR由2条高度可变肽链组成异二聚体,包括TCR2(α/β)和TCR1(γ/δ)。由于携带αβ肽链的T细胞占了绝大多数(98%),所以接下来提及的TCR都默认为TCR2(α/β)。TCRα链和β链均各有1个可变抗原结合区、1个恒定区和1个跨膜结构域。但TCR链并不具备独立传递信号的能力,这是由于TCR的2条肽链(αβ)胞内区很短,无法独立传递信号(见图1)。这种结构特性使得其必须通过跨膜区(带正电荷)与分化簇3(cluster of differentiation 3,CD3)分子带负电的跨膜区非共价结合,形成TCR-CD3复合物,进而将TCR识别抗原的信号传递到胞内,将抗原特异性识别与效应反应结合起来,启动一系列信号反应。TCR通过1个抗原识别亚基(TCRαβ)和3个CD3信号亚基(CD3δε,CD3γε和CD3ζζ)形成八聚体,而CD3γ,CD3δ和CD3ε链各含有1个免疫受体酪氨酸活化基序(immunoreceptor tyrosinebased activation motif,ITAM)基序,CD3ζ链含有3个ITAM基序,因此1个TCR-CD3复合物共含有10个ITAM基序和20个关键的酪氨酸磷酸化位点[1](见图1)。这些ITAM基序中的酪氨酸磷酸化在TCR信号转导过程中发挥重要作用。2019年,Dong等[2]报道了配体未结合状态下人TCR-CD3复合物的冷冻电镜(cryo-EM)结构,分辨率为3.7 Å,并以TCRαβ/CD3δε/CD3γε/CD3ζζ的化学计量1∶1∶1∶1进行组装,这与生化实验所得结果相呼应,进一步证实了TCR-CD3复合物的结构组成。
与TCR-CD3复 合 物 相比,1个CAR分 子通常由1个包含轻重链可变区域的单链可变区片段(single-chain variable fragment,scFv)抗体的胞外抗原识别结构域、跨膜区和胞内区组成(见图1)。CAR的跨膜区与T细胞的跨膜区功能一样,具有锚定作用,可以将CAR固定在T细胞膜上,使其稳定地发挥信号转导功能。CAR的胞内区则由1个(二代CAR)或2个(三代CAR)共刺激受体和CD3ζ胞内结构域构成。与TCR一样,CAR信号序列上同样具有多个ITAM,能将CAR接收到的抗原信号转化为活化信号,激活T细胞,使其增殖并分化为效应性的T细胞,发挥抗肿瘤等免疫应答功效。但是与天然的TCR相比,CAR仅含有3个ITAM。一项对小鼠TCR信号转导的研究表明,低多样性的ITAM足以诱导细胞因子的产生,但TCR驱动的细胞增殖需要高多样性ITAM,从而揭示了TCR信号转导具有分级性质[3]。此外,更大的ITAM多样性能够介导更强的下游信号转导[4],以确保更高比例的细胞在抗原刺激下被激活,而不是简单地放大单个T细胞的信号强度。因此,合适的ITAM数量也许在T细胞功能发挥中具有重要作用。表1对TCR与CAR的特性做了比较。

表1 T细胞受体和嵌合抗原受体的特性对比Table 1 Comparison of characteristics between T cell receptor and chimeric antigen receptor
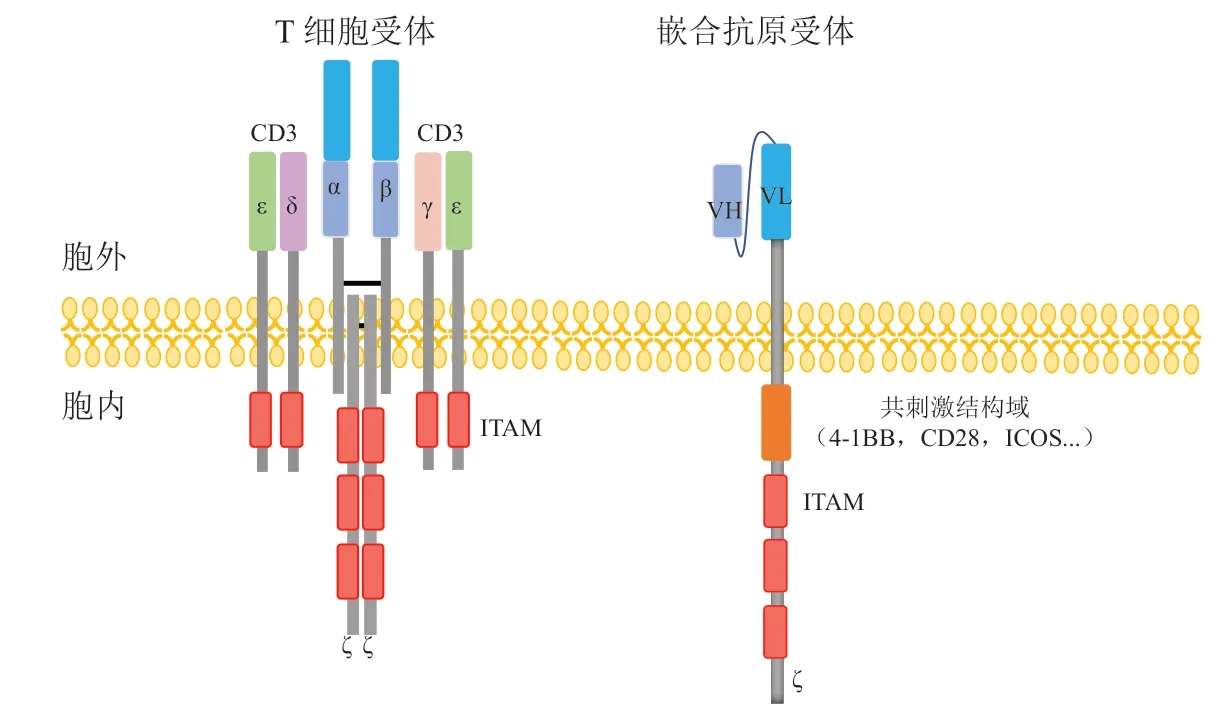
图1 T细胞受体和嵌合抗原受体的结构示意图Figure 1 Structure diagrams of T cell receptor and chimeric antigen receptor
2 T细胞受体的抗原识别
TCR和CAR识别的抗原类型不同。TCR能够识别由主要组织相容性复合物(major histocompatibility complex,MHC)呈递的抗原,CAR则利用其scFv识别细胞表面分子。目前的生物信息学预测表明,大约27%的人类蛋白质组包含膜整合结构,这些结构可能在细胞表面表达,因此这些蛋白质能够被CAR识别[12-13]。这同时也意味着有很大一部分表达在癌细胞内的潜在靶点因不能被CAR有效识别而不适用于CAR-T疗法。相比之下,TCR可以识别来自整个蛋白质组的抗原表位,包括位于细胞表面、细胞质和细胞核内的靶标分子。因此,相对于CAR,TCR可以识别更为广泛的抗原类型,不仅仅局限于细胞表面分子。这也许正是TCR-T在治疗实体瘤方面具有更好疗效的原因之一。
TCR和CAR对抗原的亲和力不同。在工程化的T细胞中,TCR对抗原的亲和力至关重要,能够决定T细胞治疗的安全性和有效性[6]。使用scFv识别域的CAR对抗原的亲和力通常在纳摩尔(nmol · L-1)级别[5]。相比之下,天然TCR的亲和力的数量级通常在微摩尔(μmol · L-1)级别[14]。基于此,许多学术界和工业界研究人员想利用CAR-T对抗原的高亲和性来获得良好的抗肿瘤功效。然而许多实验结果显示,与高亲和力的CAR相比,具备低亲和力CAR结构的CAR-T显示出更强的增殖能力和抗凋亡等持续性和抗肿瘤能力[15-16]。因此,抗原亲和力是否与抗肿瘤功能呈现正相关仍是一个有待商榷的问题。
TCR和CAR在抗原识别的敏感性方面也不尽相同。虽然基于scFv的CAR相对于TCR对其目标抗原具有更高的内在亲和力,但TCR-T可以感受并响应靶细胞上更少的分子,即能够对极少的抗原肽-MHC复合物(peptide-major histocompatibility complex,pMHC)产生反应,甚至单一抗原pMHC即可将其激活[17]。相反,CAR修饰的T细胞需要高好几倍数量级的抗原刺激才能实现与TCR-T同等水平的细胞因子释放。Harris等[18]通过将传统的α/β异二聚体或CAR与CAR胞内信号基序结合,比较了TCR和CAR在相同靶抗原的刺激下两者信号的差异,结果表明尽管TCR在膜表面的水平低于CAR,但TCR信号对抗原的敏感度是CAR信号的10 ~ 100倍;另外,定量的单分子活细胞成像结果显示TCR的敏感性是CAR的1 000倍[7]。这种敏感性数量级上的差异的内在机制尚不完全清楚,其中一种解释为相对低亲和力的TCR比相对高亲和力的scFv拥有更快的抗原解离速率(off-rate),因而能够连续不断地与单个pMHC进行反应从而扩大下游信号[19]。这种快速的抗原识别可能对实体瘤的杀伤更有利。
3 T细胞受体的信号启动
尽管关于TCR信号转导的机制方面的研究层出不穷,但TCR的触发机制仍不完全清楚,目前主要有以下几种机制(不一定相互排斥)被提出。
3.1 动态隔离模型
当TCR复合物与pMHC复合物结合后,会与抗原提呈细胞之间形成免疫突触。动态隔离模型(kinetic segregation model)可描述为在免疫突触中,TCR处于中心位置并形成“紧密结合区域”(close contact zones),进而排斥一些大分子蛋白,如磷酸酶CD45分子[20]。这种机制最终导致的结果即为TCR/CD3近端的激酶和磷酸酶平衡失调,从而使得CD3被磷酸化,促进T细胞活化和信号转导。在CAR与靶细胞特异性表达抗原结合的表面,CD45同样处于被排除状态以促进信号转导。因此,动态隔离模型在CAR信号中也许同样重要[21]。
3.2 动力学校对机制
动力学校对机制(kinetic proofreading mechanism)是指pMHC和不同的TCR相互作用过程中,不停地结合和解离,进行动力学校对;当TCR与pMHC相互作用的半衰期(t1/2)可以维持足够长的时间,便可以引发下游信号事件的发生。近年来利用光遗传学技术的研究也支持TCR的动力学校对机制,该项研究利用光来调控TCR或CAR与抗原的相互作用时间。研究表明,CAR具有类似于TCR的校对机制,因此TCR与CAR仅在其以足够的强度与配体结合时才响应,以确保有足够的时间与抗原接触[22]。这种可能性对于优化CAR设计的灵敏度和特异性具有重要意义。
3.3 串行参与模型
串行参与模型(serial engagement model)常与动力学校对机制并列提及。该模型阐述TCR与pMHC达成紧密结合就像接力一样,即单个pMHC可与多个TCR进行连续性的接触,而这些连续性接触积累起来的结果,使得TCR与pMHC达到紧密结合,最终导致T细胞的完全活化[23-24]。
3.4 构象形变模型
TCR属于精巧组装的复合物。构象形变模型指出,pMHC的刺激会引起TCR分子产生构象形变,而微小的构象改变都可引发不同的功能。TCR的构象改变发生在其胞外区、跨膜区和胞内区。越来越多的证据表明,TCR作为机械传感器,当相应配体与其特异性结合后,TCR-CD3复合物在其胞外区发生依赖于力的结构改变[25]。这种机械力来源于细胞外和细胞内。关于细胞外机械力(external force),一方面,位于细胞表面的TCR能够受到血液剪切力的影响;另一方面,细胞极化会使得T细胞接收来自细胞前沿产生机械外力[25]。另外,在T细胞与抗原呈递细胞(antigen presenting cell,APC)或靶细胞形成免疫突触时,细胞骨架重组导致的逆行流动则会对TCR施加大量内在机械力(internal force)[26]。TCR可以利用机械力引发pMHC构象变化,多步级联放大激动型和非激动型抗原的差别,从而在面临多样性的抗原时,保持与特定pMHC紧密的锚定,实现精准的抗原识别[27]。对于CAR而言,目前尚不清楚是否存在同样的机制。但是,CAR分子结构是支持其进行力依赖的分子内结构改变的。TCR跨膜区的构象改变涉及CD3ζ,TCRα以及TCRβ跨膜区。CAR的跨膜区由CD8α或CD28的跨膜区构成,目前关于CAR跨膜区构象形变引发其功能改变的研究尚未见报道。
在TCR-CD3复 合 物 中,CD3ε的 胞 内 区 含有1个富含碱性氨基酸的区域(basic residue rich sequence,BRS)、1个富含脯氨酸的区域(proline rich sequence,PRS)和1个ITAM;CD3ζ的胞内区含有3个ITAM,以及2个夹在其间的BRS;CD3γ和CD3δ的胞内区各含1个ITAM。研究表明,带正电的CD3ε和CD3ζ的BRS可以与质膜内叶中带负电的酸性磷脂相结合,暗示两者能够在pMHC结合时进行动态的结合与释放,并且这种动态化的变化与CD3ε和CD3ζ的ITAM基序的磷酸化机制密切相关。在T细胞静息状态下,CD3ε的BRS区域可以与细胞质膜上的酸性磷脂发生静电相互作用,从而使得整个CD3ε的胞内区被屏蔽在膜脂双层中。这种膜屏蔽机制既屏蔽了CD3ε的酪氨酸位点,同时也阻止了BRS区与淋巴细胞特异性蛋白酪氨酸激酶(lymphocyte-specific protein tyrosine kinase,Lck)的相互作用,达到抑制TCR整体磷酸化的效果[28]。当TCR与特异性的抗原肽-MHC复合物结合后,会使得TCR发生形变,CD3ε胞内区从膜上解离下来,暴露其中的酪氨酸磷酸化位点。此外,由于T细胞活化导致细胞内钙离子浓度的上升,内流的钙离子可以通过中和酸性磷脂负电荷的方式使得未直接接触抗原的TCR中的CD3ε胞内区从膜上解离[29];于是,暴露于胞质的游离CD3ε ITAM进一步被下游的蛋白质酪氨酸激酶磷酸化,进而发生一系列磷酸化级联反应,从而放大TCR活化信号。另外,还有学者指出pMHC与TCR的结合会暴露CD3ε的PRS区域,这一暴露的PRS区域可以为含有SH3结构域的非催化域酪氨酸激酶(non-catalytic region of tyrosine kinase,Nck)提供结合位点并进一步放大TCR信号[30-31]。但不管是BRS还是PRS,都暗示TCR信号转导不是简单的磷酸化级联反应,其中必定存在尚不清楚的复杂结构变化,因此,对pMHC与TCR结合后,导致TCR复合物磷酸化及其结构变化的理解仍然是一个非常大的挑战。近期以机械力为参数的研究另辟蹊径,利用TCR与激动型pMHC配体的结合具有机械力依赖性的特点,通过观测TCR与pMHC配体结合时形成“捕获键(catch bond)”还是“滑移键(slip bond)”,研究人员能够推测下游信号激活的差异[32]。
受体聚集也是导致构象形变的机制之一。配体结合会导致膜蛋白的聚集或构象改变。Janeway于1995年首次提出TCR的活化伴随TCR的聚集和构象改变。由于常规T细胞的激活并不需要pMHC多聚体[33],因此受体聚集可能并不是TCR激活的主要机制。而CAR启动信号传递必须依赖CAR蛋白分子聚集[34-35],因此这种机制可能在CAR中发挥更为关键的作用。
4 T细胞受体和嵌合抗原受体免疫突触的形成
上文提到,TCR和CAR信号转导的起始伴随受体聚集。对于天然TCR而言,当TCR与pMHC结合后,T细胞和APC之间会形成复杂而有序的超分子结构,称为超分子激活聚集体(supramolecular activation cluster,SMAC),即免疫突触。这种细胞与细胞之间的接触包括“immunological kinapse”(IK)和“immunological synapse”(IS)。其中kinapse是TCR在受体迁移寻找相应配体pMHC的过程中形成的一种瞬时且不稳定的结构,synapse则是TCR与pMHC识别后形成的稳定长时间接触面[36]。TCR刺激和共刺激信号水平、T细胞的分化状态以及靶细胞的表型共同决定了细胞是否形成对称、稳定的IS或瞬时的IK,或在2种模式之间过渡[37]。IS由3种不同类型的受体组成,包括TCR、黏附受体和共刺激或共抑制受体,以及它们相关的信号转导分子。在TCR与pMHC识别过程中,T细胞和APC之间会形成约为15 nm的间隙,这为受体和配体形成IS提供了空间[38]。黏附分子通过将细胞膜紧密连接在一起来支持IS的结构,这对于持续的抗原识别必不可少。另外,IS招募的共刺激受体或共抑制受体可以显著调节T细胞的激活。T细胞与B细胞或平面抗原呈递底物相互作用的免疫荧光成像显示,SMAC由3个同心圆环组成,类似于“牛眼(bull,s eye)”[39](见图2)。中央SMAC(cSMAC)形成中心环,外围SMAC(pSMAC)和远端SMAC(dSMAC)在其外围环绕。每个环都有自己特殊的组成和功能。cSMAC包含一定浓度的TCR和共刺激分子CD28,负责伴随突触形成的关键T细胞激活信号事件;pSMAC包含一系列黏附分子,如淋巴细胞功能相关抗原-1(lymphocyte functionassociated antigen-1,LFA-1),负责稳定细胞间的相互作用;dSMAC由丝状肌动蛋白组成,有助于对突触施加机械力,另外,dSAMC还聚集许多酪氨酸磷酸酶,最典型的即为CD45[40]。
CAR-T突触并不像SMAC那样具有高度组织性。相反,它们是杂乱无章的、零散的信号簇,缺乏明确的结构。除了不形成较小且不典型的F-肌动蛋白环之外,CAR形成的免疫突触缺乏黏附环并且Lck激酶簇呈散乱分布[9,41](见图2)。因此,CAR-T与其靶细胞形成的免疫突触在结构上不同于经典免疫突触[9,21]。这也可能是两者具有杀伤能力差异的重要原因之一。

图2 T细胞受体和嵌合抗原受体免疫突触的比较Figure 2 Comparison of immune synapses between T cell receptor and chimeric antigen receptor
CAR-T突触和经典pMHC/TCR突触之间的这些结构差异会导致功能差异。对突触形成后TCR-T介导的杀伤与CAR-T介导的杀伤的时程比较显示,CAR-T的信号强度比TCR-T信号更强且上升更快,穿孔素(perforin)和颗粒酶B(granzyme B)的释放更快,脱落也明显更快。因此,与细胞毒性T淋巴细胞(cytotoxic T lymphocyte, CTL)相比,CAR-T杀死靶细胞的速度更快[9,41]。而无序的CAR-T突触可能就是其快速杀伤活性的结构基础[42]。需要指出的是,这里CAR-T的快速杀伤活性建立在高抗原密度的基础上。在低抗原密度下,CAR-T近端信号弱且常受限于其活化阈值而不能够对靶细胞进行有效杀伤[7]。这种对靶抗原的低敏感性正好能够解释在CAR-T免疫治疗中由于肿瘤抗原低表达或无法检测而导致癌症复发的现象[43]。
了解突触的形成机制并鉴定其质量非常有必要。基于玻璃制成的平面脂质双层系统的成像系统已被用于预测IS的质量,并证明IS的质量与CAR-T功能相关联[44]。更有数据表明,在病理条件下,高IK频率表明T细胞无能化,进入免疫耐受状态而阻止肿瘤免疫[45]。由此可见,合适的IS形成是T细胞抗肿瘤功能发挥的必要条件。然而,目前IS在T细胞中的形成机制仍未完全了解,还有许多关键问题有待回答。例如CAR-T这种无序IS结构产生的机制尚不清楚。有研究人员将具有高亲和力识别结构的CAR与天然的TCR融合或连接后发现,改造后的T细胞能够与相应的靶细胞形成典型的稳定的IS结构[46-47]。因此,这种IS结构的混乱也许并不是由于CAR对抗原的高亲和力及它的非MHC限制性性质所导致。鉴于IS结构的重要性,仍需进一步了解其在抗肿瘤作用中的地位。
5 T细胞受体的近端信号转导
TCR通过其配体识别亚基与APC上特异性表达的pMHC复合物结合,激活TCR信号通路。SRC家族蛋白酪氨酸激酶Lck是TCR信号起始的关键因子,它能够与CD4和CD8共受体的胞质结构域结合,并分别通过CD8或CD4与MHC I类或MHC II类复合物的共结合募集至TCR。处于活化构象的Lck可以磷酸化ITAM,每个ITAM具有2个可被磷酸化的关键酪氨酸残基,被双磷酸化的ITAM可以招募相对分子质量为70 000的ζ链相关蛋白激酶(zeta-chain-associated protein kinase-70,ZAP-70),使蛋白酪氨酸激酶ZAP-70结合到CD3链中[48]。这里需要说明的是,Lck激酶包括游离状态和与共受体偶联的2类群体,它们均能在TCR活化后磷酸化CD3,但游离状态的激活态Lck会被更早地招募至TCR起始信号传递[49]。换言之,由Lck介导的TCR活化可以分为2个阶段:首先,游离态Lck激酶最先被招募到TCR并磷酸化CD3 ITAM;接着,与共受体偶联的Lck被招募,一方面能够促进TCR与pMHC的结合,另一方面起到放大信号的作用[49-51]。随后,与ITAM结合的ZAP-70被磷酸化并招募更多的ZAP-70分子且进一步传递信号。由此可见,Lck和ZAP-70之间存在着非常严格的层级关系,Lck位于ZAP-70的上游[24]。
Lck在TCR的信号转导起始中发挥非常重要的功能,它将细胞外TCR与pMHC的结合事件与细胞内生物化学事件联系在一起。研究发现,Lck的定位和构象变化是TCR信号转导的重要决定因素[52-53]。Lck由1个UD结构域(unique domain)、2个调节结构域(SH3和SH2)和1个激酶结构域(kinase domain)组成,它包含若干个磷酸化位点,其激酶活性受2个关键调节性酪氨酸位点Y394和Y505的磷酸化和去磷酸化控制。Y394位于Lck激酶环中,其自磷酸化可以稳定催化结构域的活性构象。而C末端Src激酶(C-terminal Src kinase, Csk)能够对Lck的活性负向调控,它通过对Lck羧基末端Y505残基的磷酸化,促进Y505与Lck SH2结构域的分子相互作用,从而稳定了使催化结构域失活的折叠构象。CD3ε ITAM存在部分单磷酸化现象,这种单磷酸化的CD3ε能够招募抑制性激酶Csk,发挥抑制性作用[54]。另外,跨膜酪氨酸磷酸酶CD45可使Lck磷酸化的Y505和Y394去磷酸化,因此,CD45可以直接调节控制Lck活性。上文中已经提到,在T细胞活化早期,游离的活化型Lck最先被招募至TCR附近磷酸化CD3链以及一些关键的激酶来起始T细胞活化,这可能是由于游离的Lck比与共受体结合的Lck具有更高的流动性、更多的Y394磷酸化以及更强的激酶活性[55],因而游离的Lck对于TCR的早期活化非常重要;但当共受体结合的Lck不能被招募到TCR周围时,T细胞同样不能被有效地活化[56]。另外,作为早期活化的重要成分,ITAM和ZAP-70的磷酸化都需要具有活性的Lck的存在,因此活化型的Lck必须位于TCR:pMHC附近足够长的时间,以确保这些事件的发生。综上,Lck的定位和构象变化都将影响免疫突触的形成和稳定性。
关于CAR,与TCR共享相同的CD3ζ胞内区的同时,它还包含CD28和肿瘤坏死因子超家族成员4-1BB等共刺激结构域。因此CAR与TCR之间,以及包含不同共刺激结构域的CAR之间的近端信号传导不尽相同。例如,在包含CD28或4-1BB的2种二代CAR中,配体结合后,CD3ζ ITAM的磷酸化虽然位置相同,但强度不同,包含CD28的CAR在早期时间点检测到更强的磷酸化信号。这可能是因为CD28含有1个可以结合Lck的脯氨酸富含区,使其能够像共受体一样招募Lck[57]。有关CAR的近端信号传导仍属于活跃的研究领域。
6 T细胞受体的下游信号转导
在上述提及的一系列酪氨酸激酶初始激活后,T细胞活化衔接因子(the linker of active T cells, LAT)和含SH2结构域相对分子质量为76 000的白细胞磷酸化蛋白(SH2 domain-containing leukocyte phosphoprotein of 76 000, SLP-76)被磷酸化,活化T细胞,募集多个衔接因子和效应分子,形成LAT信号转导体来调节下游信号[58]。LAT的磷酸化由ZAP-70介导,而两者的连接通过Lck激酶的SH2作为桥梁所介导[59]。随后,磷酸化的LAT招募磷脂酶C-γ(phospholipase C-γ, PLC-γ)、生长因子受体结合蛋白2(growth factor receptor-bound protein 2, GRB 2)和Shc下游GRB相关接头蛋白(GRB-related adaptor downstream of Shc, Gads)。LAT相关效应分子的活化导致信号通过3种主要信号通路进行转导:钙调蛋白、丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)和核因子κ-B(nuclear factor kappa-B,NF-κB)信号通路。钙调蛋白信号转导会导致活化T细胞核因子(nuclear factor of activated T cells,NFAT)发生核转移。MAPK信号转导导致肌动蛋白聚合以及转录因子FOS,JUN和激活蛋白-1(activator protein-1,AP-1)的激活;NF-κB信号转导使REL和NF-κB转录因子核移位;这些转录因子协同作用引起T细胞增殖和迁移、细胞因子分泌、免疫突触的形成以及相关效应功能。由此可见,LAT是T细胞信号转导的必需支架蛋白。
由于CAR是利用CD3ζ作为信号基序去激活T细胞的效应功能,因此普遍认为CAR存在与TCR相同的信号转导机制。许多着眼于CAR下游的磷酸化事件的研究结果显示CAR-T和TCR-T的磷酸化事件虽然在量级和动力学上存在差异,但CAR-T中并未出现新的通路[57,60-61]。CAR利用相同的分子机制去转导信号,包括PLC-γ,ZAP-70,LAT和Src家族蛋白激酶等。但也有研究表明,CAR和TCR拥有不同的信号通路,即CAR的信号转导可以绕过LAT[10]。此外,CAR-T对抗原刺激之后的表现仍有无法解释的方面。例如,与TCR相比,CAR触发下游信号分子的酪氨酸磷酸化更快,细胞毒性颗粒释放更快[62]。通过直接将TCR和CAR表达在同一细胞中进而比较它们的信号过程,发现尽管TCR信号与CAR信号相比更缓慢和更微弱,但TCR信号更为持久[9]。另外,两者对抗原的敏感性不同。已有许多研究指出,TCR的信号转导具有高度的敏感性,在只有单个抗原pMHC存在的情况下,就足以激活T细胞反应[8,63]。然而,CAR的激活需要更多的抗原分子刺激[62]。有学者解释这种低敏感性是由于CAR活化时近端信号明显减弱导致,尽管CAR在免疫突触内的抗原结合亲和力方面表现优于TCR,但其胞内区CD3ζ链的酪氨酸蛋白激酶ZAP-70的招募效率很低[7]。CAR的这种信号转导缺陷,一方面限制了CAR-T疗法治疗表面抗原密度低的肿瘤的疗效;另一方面,这也许解释了CAR-T临床治疗中患者由于肿瘤的抗原表达水平低或无法检测到而出现癌症复发的现象。
CAR和TCR在激活后具有许多相似的下游信号事件,如它们都分泌穿孔素和颗粒酶B去诱导靶细胞的凋亡,并表达细胞因子和趋化因子来增强免疫功能或调控炎症反应,然而,临床报告却显示出不同的结果。细胞因子释放综合征(cytokine release syndrome,CRS)表现为CAR-T疗法中最常见的毒性,其确切的机制尚不完全清楚;其潜在的原因可能是CAR异常或过度活化的信号。在静息态T细胞中,CD3ζ肽链对借助于其C末端的正电与质膜上带负电的酸性磷脂相互作用,从而达到屏蔽信号的作用[29]。而当抗原不存在时,CAR-T仍存在很强的基底信号[64]。因此,高强度的基底信号也许放大了信号而促进细胞因子的产生,进而进一步导致细胞因子风暴的产生而影响CAR-T疗法的疗效。此外,在天然的TCR复合物中,CD3ε ITAM可被单磷酸化,从而招募Csk磷酸酶,发挥负调控功能[54]。提示除了过强的活化信号,有关负调控信号在CAR中的缺失也可能是导致CRS的重要原因。总之,天然TCR复合物结构远比想象的更复杂,故应当意识到其在指导过继性免疫疗法中T细胞设计改造方面的重要性。
7 结语
TCR-T和CAR-T疗法均为过继性免疫疗法治疗癌症的前沿技术,且基本思路均为将T细胞在体外进行修饰、扩增后再植入患者体内,2种改造过的受体都能够介导T细胞发挥肿瘤杀伤作用。但两者显然存在许多不同:包括结构、抗原识别机制和识别类型、对抗原响应的敏感度等。CAR-T疗法已在难治性白血病和淋巴瘤患者中诱导了显著的反应,并获得了美国FDA的批准,因此其对血液瘤及淋巴瘤的功效毋庸置疑。但目前许多临床数据均显示TCR-T疗法在治疗实体瘤的能力方面比CAR-T疗法略胜一筹。总的来说,从2种细胞本身特点出发:首先,CAR-T靶向肿瘤抗原,而TCR-T除了识别表面抗原之外,还能够通过白细胞抗原(human leukocyte antigen, HLA)提呈进而靶向细胞内蛋白质组,这意味着TCR-T可以识别广泛的肿瘤抗原,不限于膜表达抗原,还包括一些由体细胞突变产生的新抗原(neo-antigen)。此外,从患者体内取出的T细胞所制备的TCR-T与患者HLA等位基因匹配,避免了移植后的免疫排斥反应。最后,TCR-T有更好的肿瘤浸润和分布,而CAR-T由于识别表面抗原倾向于分布在肿瘤周围,使得TCR-T在治疗实体瘤时更有效[11]。尽管如此,目前对于特异性肿瘤抗原靶点的发现还甚少,并且从病人体内取出的T细胞无法在体外有效地大量扩增,体外的制备工序费时费力。另外,科学家们现在还无法很好地解决T细胞疗法治疗后的大量不良反应。综上所述,过继性免疫细胞疗法治愈癌症任重而道远。不论是CAR-T疗法,还是TCR-T疗法都仍存在一系列问题,因此很有必要清楚理解基本的TCR信号转导机制,并为设计更好的T细胞产品提出新的策略,以求其在实体瘤临床治疗中得到进一步应用。
猜你喜欢
保健与生活(2022年5期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
皮肤病与性病(2021年3期)2021-07-30
科学24小时(2018年1期)2018-01-10
天津农业科学(2017年8期)2017-08-11
吉林农业(2017年5期)2017-05-13
现代养生·下半月(2016年6期)2016-10-21
恋爱婚姻家庭·青春(2016年10期)2016-10-10
江苏农业科学(2015年5期)2015-10-20
