
一类新型含平面四配位碳、硅和锗的化合物的结构与成键性质
2021-11-08 12:40张彩云张于微曹泽星
厦门大学学报(自然科学版) 2021年5期
张彩云,方 磊,张于微,曹泽星
(厦门大学化学化工学院,福建省理论与计算化学重点实验室,福建 厦门 361005)
自1874年van’t Hoff[1]提出C原子的四面体结构以来,四面体构型已成为C原子成键的经典模型.1960年,Pauling[2]提出C原子采取sp3杂化的方式与周围原子成键,从理论上解释了碳四面体结构的成因.Monkhorst[3]在讨论非对称C原子四面体结构互变时,首次假设平面四配位碳(planar tetracoordinate carbon, ptC)构型为其非断键异构化的过渡态.ptC概念的出现在理论和实验上引起了人们广泛的研究兴趣.1970年,Hoffmann等[4]基于近似计算,分析了具有D4h对称性的平面甲烷分子的成键性质.他们认为,C原子采用sp2杂化与周围4个H原子形成2个正常的σ-型的C—H键和1个三中心两电子(3c-2e)键,最高占据分子轨道(highest occupied molecular orbit,HOMO)为垂直于分子平面π-型的pz轨道,最低未占分子轨道(lowest unoccupied molecular orbit,LUMO)具有σ反键特征,不同价键结构间的共振导致4个C—H键等价.基于这一成键特点提出了稳定含ptC化合物的策略,即把连接C的H原子替换为σ给电子效应或π吸电子效应的取代基以稳定C上的pz孤对电子.1976年,Collins等[5]基于半经验和从头算理论计算,在理论上成功设计了含ptC的分子.1977年,Cotton等[6]首次实验合成含ptC的化合物.随着更多含ptC分子的成功制备[7-10],平面高配位碳体系也受到关注[11-12].这些碳的平面配位结构作为低维纳米材料的基本单元,可用于功能材料的设计[13-15].
C、Si和Ge均为同族元素,和C相比,平面四配位硅和锗(ptSi和ptGe)的研究相对较少,主要集中在一些平面四配位的金属团簇体系[16-18].最近,Sun等[19]在理论上对一些含ptSi化合物和二维单层纳米片进行了计算模拟研究,结果表明含ptSi的二维材料具有非常好的储锂性能.1979年,Würthwein等[20]分析了含ptSi与ptC分子的成键性质,发现不同于含ptC的分子, 含ptSi分子的HOMO和LUMO分别为σ-型和π-型轨道.因此,稳定其平面四配位结构需连接强的π电子给体或σ电子受体.2000年,Boldyrev等[16]首次在实验中观测到含ptSi的团簇SiAl4-(17e-)和SiAl4(16e-).2012年,Alexandrova等[21]提出这些团簇的几何结构源于体系芳香性与共价性的竞争,相对强的芳香性有利于平面构型.理论上还预测了一系列价电子数为18(SiAl42-、SiIn42-和SiC2Li2)和14(HSiAl3、Ca3SiAl-和Mg4Si2-)的团簇[22].
2018年,实验上成功合成了第一例含ptSi的非金属配体分子:杯[4]吡咯氢化硅酸盐[23].不同于其他超配位氢化硅阴离子,含ptSi的阴离子[H--SiN4]在水和空气环境中是稳定的,不具有氢化物反应性.为了探明这类体系的成键特征,揭示平面四配位构型稳定性的调控因素,本研究构建了系列含ptC、ptSi和ptGe的分子,并应用密度泛函理论对其结构、稳定性及谱学性质进行了系统的计算研究.
1 计算模型与方法
基于杯[4]吡咯氢化硅阴离子的中心骨架结构,构建了平面四配位C20H16Y4X和对应的四面体结构C16H16Y4X模型,分别记为ptX-Y和ttX-Y(X=C,Si,Ge;Y=B,N),如图1所示.其中,杯[4]吡咯氢化硅阴离子体系中的甲基用H原子替代.测试计算表明,这种结构简化不影响中心平面四配位结构和成键特征.应用Gaussian 2009软件[24],在B3LYP/6-311G(d,p)水平下,对ptX-Y和ttX-Y两类体系进行几何结构优化和频率计算.对于ptX-Y结构模型,获得了4种稳定的含平面四配位结构单元的分子:C20H16B4C、C20H16N4Si、C20H16N4Ge和C20H16B4Ge(分别记为ptC-B、ptSi-N、ptGe-N和ptGe-B).为了理解平面化的稳定因素及成键特征,进行自然价键轨道(natural bond orbital,NBO)分析[25].此外,应用ChemCraft软件[26]对分子轨道可视化,并使用Multiwfn程序[27]对特殊轨道进行定域化分析、组成成分分析[28]和Fuzzy键级计算.基于优化的稳定结构,在TD-B3LYP/6-31+G(d,p)水平下,预测了这4种平面四配位分子的电子吸收光谱.
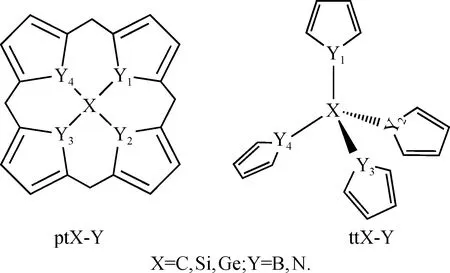
图1 平面四配位ptX-Y和四面体ttX-Y的分子结构模型
2 结果与讨论
2.1 结构与稳定性


键长单位为pm;Φ1为Y1—Y2—Y3—Y4的二面角,Φ2为Y1—Y2—X—Y3的二面角.
2.2 分子轨道与成键特征
ttX-Y分子的成键特征十分清晰,NBO计算结果显示,X原子采取sp3杂化形成经典的四面体结构.为探究ptX-Y分子平面化和稳定性的原因,对其特定前线分子轨道进行分析.对于ptC-B化合物,C原子的价层轨道对多条分子轨道(HOMO-4~HOMO-50)均有贡献.为了更直观地理解其成键特征,对分子轨道进行定域化,如图3所示,可以看出C原子可以与周围B1、B4原子或B3、B4原子形成2个3c-2e的σ键,轨道成分计算显示这2个σ键中C原子的主要贡献轨道为2s轨道和2px或2py轨道.此外,C原子还能和周围1个B原子形成1个近似2c-2e的σ键,其中C原子的2s、2px和2py的组分占比分别为14.13%,19.92%和21.40%.ptC-B在形成多中心两电子σ键的过程中共有6个电子参与,其中4个B原子各提供1个电子,C原子提供2个电子,C原子剩下的2个电子则分布在2pz轨道上,并与周围B原子的pz空轨道形成共轭π键.这里的C—B成键无论是在σ空间还是在π空间均有明显的离域特征,5c-6e的面内离域σ键和5c-2e离域π键与Hoffmann等[4]提出的ptC-H4分子中平面碳的成键图像并不完全相同.
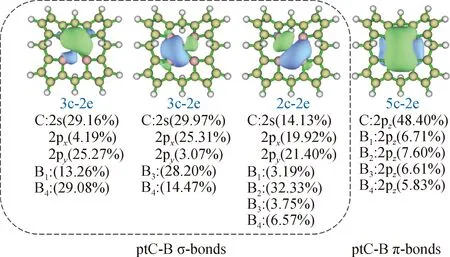
图3 ptC-B分子的定域化分子轨道(等密度面值:0.03)
图4(a)给出了ptSi-N分子几个重要的分子轨道图,其分子轨道组成显示,ptSi-N中Si原子的3s、3px、3py和3dxy轨道分别与N原子形成σ键,相应的分子轨道占比分别为6.70%,8.59%,8.59% 和5.88%,中心Si原子近似为sp2d杂化.NBO分析证实ptSi采取sp2d杂化的方式与周围4个N原子成键,而Si原子剩余的3pz空轨道与周围N原子的pz轨道形成共轭π键,如图4(a)中的HOMO-7轨道所示.和同族C元素相比,Si原子具有3d价轨道和相对稍小的3p-3d能级差,导致其在平面四配位结构中形成不同的成键性质.显然,中心Si原子的3pz空轨道的存在及其稳定化导致sp2d杂化并形成稳定的平面四配位结构.同时,发现ptGe-N的成键模式与ptSi-N基本一致,相应的分子轨道见图4(b).
结构优化和振动频率计算揭示,含ptGe的ptGe-B分子是稳定的.图4(c)给出了与ptGe-B结构单元对应的成键分子轨道,不同于ptGe-N的分子轨道,ptGe-B与ptGe-N平面四配位结构单元具有不同的成键方式.从图4(c)可以看出:Ge原子的4s、4px和4py轨道参与形成3个σ键,占比分别为5.90%,17.05%和17.04%,其中4s轨道的贡献相对较小;Ge原子的px和py轨道直接与周围B原子形成2个多中心两电子的σ键.和ptC-B分子类似,Ge的4pz轨道上的孤对电子与周围B原子的pz空轨道形成5c-2e的离域π键.尽管ptGe-B和ptC-B分子存在相同的离域π键,但由于Ge具有较大的s-p能级差,平面四配位GeB4结构单元的σ键成键模式和ptC却存在显著差异.
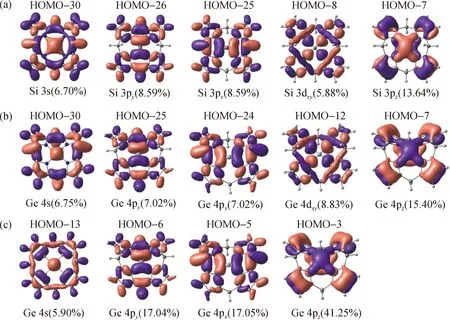
图4 ptSi-N(a)、ptGe-N(b)和ptGe-B(c)的分子轨道(等密度面值:0.03)
对于第4主族C、Si和Ge,C与B可形成含平面四配位结构单元CB4的化合物,Si与N可形成含平面四配位结构单元SiN4的化合物,而Ge可分别与B、N形成含平面四配位结构单元GeB4或GeN4的化合物,这些化合物的成键差异源于不同的价轨道及能级差[29].通常s-p能级差越大,s轨道电子越稳定,因此s轨道成键的可能性越小,而p轨道成键的可能性越大[30].由于C、Si和Ge原子的s-p能级差逐渐增大,分别为5.30,5.35 和6.40 eV,所以C原子价层的s、p轨道能级更为接近,容易杂化后成键;Ge原子的s-p能级差最大,p轨道更易直接参与成键;而Si原子的s-p能级差则处于C和Ge原子之间,既不容易杂化也不容易通过原子轨道直接成键.与C原子不同的是,Si和Ge原子价层有d轨道,价层s轨道的电子能够激发到d轨道上,形成 sp2d 杂化与周围原子成键,同时周围N原子上的孤对电子再通过共轭相互作用稳定Si原子的pz空轨道.其中,Si和Ge原子的p-d能级差分别为5.61和6.01 eV,从能量角度来看,Si原子更容易被N原子平面化.
2.3 电子吸收光谱
基于优化的基态平面四配位几何构型,在TD-B3LYP/6-31+G(d,p)水平下,预测了含ptC、ptSi和ptGe的4种稳定结构的电子吸收光谱.图5和表1分别给出了相关的吸收光谱及重要电子跃迁的垂直激发能、归属和对应的振子强度(f).如图5和表1所示,ptC-B分子能够吸收的波长范围为200~400 nm,分别在252和314 nm附近有2个强吸收峰,对应的振子强度分别为0.21和0.20.其中,在252 nm附近的吸收光谱主要由基态到较高能态的σ→π*电子激发贡献,即S0→S26和S0→S27跃迁;而在314 nm附近的吸收光谱则是由S0→S16和S0→S17等π→π*电子跃迁产生.值得注意的是,ptSi-N和ptGe-N分子的吸收光谱非常类似,均有2个主要吸收峰:ptSi-N的2个主要吸收峰分别出现在228和350 nm,对应的振子强度分别为0.03和0.20;ptGe-N的2个主要吸收峰分别出现在236和374 nm,对应的振子强度分别为0.03和0.21.相比于ptSi-N分子,ptGe-N分子的主要吸收峰均发生一定程度的红移.从图4(a)和(b)可以看出,ptSi-N和ptGe-N 2个分子的电子吸收光谱性质的相似性和它们具有相同的平面四配位成键特征一致.
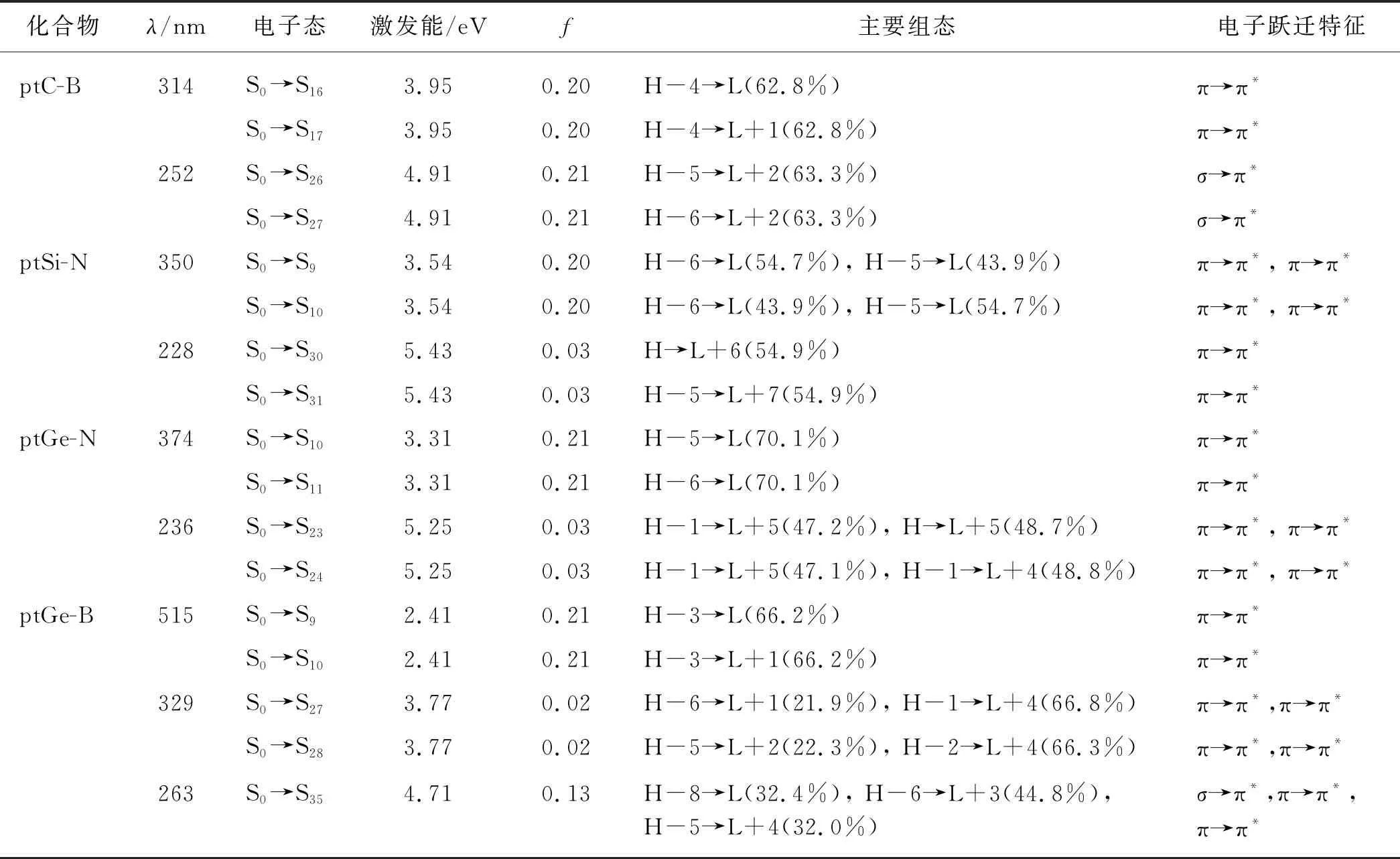
表1 平面四配位分子的电子吸收波长、电子态、垂直激发能、谐振子强度、主要组态和电子跃迁特征
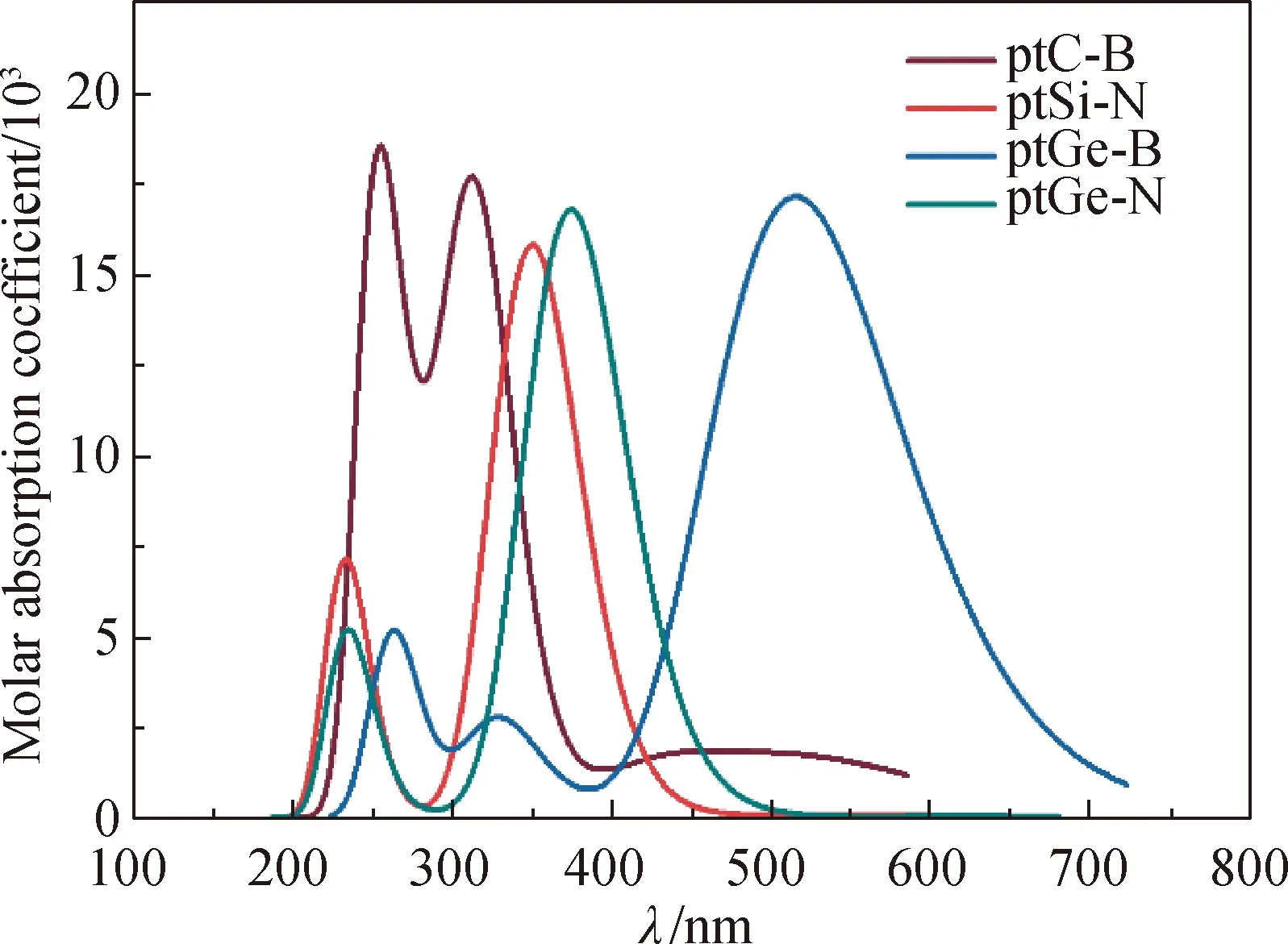
图5 4种平面四配位分子的电子吸收光谱
和以上分子体系不同,ptGe-B是唯一在可见光区内有强吸收的分子,吸收波长范围为400~750 nm,是一类潜在的可见光分子功能材料.其最强吸收位置在515 nm附近,对应的振子强度为0.21,主要来源于HOMO-3→LUMO和HOMO-3→LUMO+1的π→π*电子跃迁.此外,ptGe-B分子在相对高能区的329和263 nm附近分别有2个较弱的吸收峰,对应的振子强度分别为0.02和0.13,主要来源于π→π*电子跃迁.
3 结 论
基于实验合成的第一例含ptSi的分子结构特征,本研究从理论上设计了含ptC、ptSi、ptGe的4类化合物:ptC-B、ptSi-N、ptGe-B和ptGe-N.计算结果表明,这4类分子都具有稳定的几何和电子结构,这些平面四配位化合物中心原子与周围原子的成键强度和通常四面体结构的单键类似.由于原子价层轨道能级差的差异,C、Si和Ge原子在形成平面四配位XY4(X=C,Si,Ge; Y=B,N)结构单元中呈现出不同的成键特征.C原子s、p轨道杂化和B原子形成σ键,其pz轨道上的孤对电子与周围B原子的pz空轨道形成共轭π键.Si原子由于外层有d轨道,可以通过sp2d杂化与周围N原子形成σ键,同时N原子上pz轨道的孤对电子和Si原子的pz空轨道相互作用,形成共轭π键.Ge原子则可以同时与B和N原子形成稳定的平面四配位结构的分子.由于Ge原子的s-p能级差较大,s和p轨道难以杂化,所以Ge原子主要通过价原子轨道直接与周围B原子形成多中心化学键.这些平面四配位化合物中,ptGe-B分子的电子强吸收谱峰出现在515 nm可见光区,其余3种分子的主要吸收谱峰均在200~400 nm紫外光区,其中ptSi-N和ptGe-N分子的主要吸收谱峰的位置和强度基本类似,均位于360和230 nm附近.
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
暨南学报(哲学社会科学版)(2022年3期)2022-11-09
无机化学学报(2022年9期)2022-09-16
原子与分子物理学报(2020年5期)2020-03-17
西部探矿工程(2018年9期)2018-09-11
考试周刊(2018年39期)2018-04-19
高校招生(2017年5期)2017-06-30
电子制作(2017年1期)2017-05-17
中国塑料(2016年1期)2016-05-17
