
双环[4.3.0]环戊烯酮的合成
2021-11-02 04:59刘锟
探索科学(学术版) 2021年9期
刘 锟
宋善敏通讯作者3
1.贵州医科大学省部共建药用植物功效与利用国家重点实验室 贵州 贵阳 550014;
2.贵州省中国科学院天然产物化学重点实验室 贵州 贵阳 550014;
3.贵州农业职业学院 贵州 贵阳 551400
双环[4.3.0]结构广泛分布于萜类或生物碱类天然产物中。具有代表性天然产物有Fawcettimine[1],Palhinine A[2],Aplykurodinone-1[3,4],Alborosin[5,6],Astellatol[7]。化学结构如图1所示,它们的全合成也具有一定的挑战性,因此其合成方法有重要的研究价值。在构建[4,3,0]环结构时Pauson-Khand反应是常用的方法之一[8],但是Pauson-Khand试剂昂贵,同时较活泼而不稳定,反应剧烈。因此本研究利用廉价易得的1,4-环己二酮单乙二醇缩酮为起始原料,经过wacker氧化[9]和脱保护转化,随后以Robinson环化反应[10]合成[4,3,0]环结构。

图1 双环[4.3.0]化合物4的合成路线
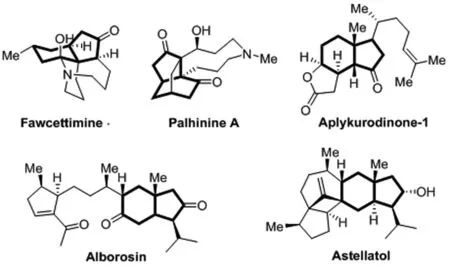
图1 含双环[4.3.0]结构代表天然产物
1 实验部分
1.1 实验试剂及仪器
集热式恒温加热磁力搅拌器(艾卡(IKA)仪器设备有限责任公司),三用紫外分析仪,旋转蒸发仪,Advance DMX400型核磁共振仪(Bruker公司,400 MHz,TMS为内标),Agilent 1100 LC/MS质谱仪,精密电子天平(RADWAG AS.X2系列分析天平)
1.2 实验部分
以1.3-环己二酮单乙二缩酮为起始原料,在LDA作用下脱质子形成羰基α位负离子与烯丙基溴取代得到化合物2,双羰基化合物3可以通过PdCl2和CuCl催化的Wacker氧化合成,随后在2 M HCl条件下脱乙二醇保护后,经后处理无需过柱纯化,粗品加入1 M KOH 溶液通过Robinson环化得双环化合物4.
化合物2合成:在500 m L 的单口圆底烧瓶中加入13 g的二异丙胺,加入60 m L干燥的THF溶液,氮气保护,转移至-78℃低温反应器中,待冷却10 min后缓慢滴加正丁基锂40 m L。滴加完后在-78℃反应15 min转移至室温20 min,完成LDA(1 M)的制备。将新制的LDA转移至-78℃低温反应器中10 min后缓慢滴加到1.3-环己二酮单乙二缩酮的THF溶液(15 g原料25溶于200 m L THF中),滴加完成后缓慢升温到-40℃,反应30 min。在该温度下加入36 g烯丙基溴,缓慢升温至室温反应10 h。TLC监控反应不在进行时加入50 m L饱和氯化铵猝灭反应,萃取水相50 m L三次,并用饱和食盐水洗涤。合并有机相,用无水硫酸钠干燥。减压蒸馏色谱柱分离纯化(石油醚:乙酸乙酯80:1~40:1)梯度洗脱,得到淡黄色油状液体17 g,收率78%。1 H NMR(400 MHz,CDCl3)δ5.74(m,J=16.8,10.2,7.8,6.4,1 H),5.05-5.01(m,2 H),4.08-3.93(m,4 H),2.74-2.61(m,2 H),2.56-2.50(m,1 H),2.38(m,J=14.3,5.1,3.2,1 H),2.11(m,J=13.3,5.8,3.5,1 H),2.07-1.93(m,3 H),1.69(t,J=13.2,1 H);13CNMR(100 MHz,CDCl )δ210.7,135.8,3 116.7,107.3,64.6,64.5,45.7,39.8,38.1,34.5,33.2;
化合物3的合成:称取500 mg PdCl2和12 g的CuCl加入到500 m L 干燥的圆底烧瓶中,加入210 m L DMF和30 m L水(DMF:H 2 O=7:1)。向反应通入氧气,搅拌1 h。用注射器加入反应物2 17 g反应16 h,TLC 监测完全反应,加100 m L水猝灭反应,用2000 m L的水除去反应溶剂DMF,用200 m L乙酸乙酯萃取3次,减压蒸馏色谱柱分离纯化(石油醚:乙酸乙酯30:1~10:1)。得到淡黄色油状液体24 g,收率94%。
1H NMR(400 MHz,DMSO)δ4.04-3.96(m,2H),3.92(m,J=5.9,2.8 Hz,2H),3.12-2.97(m,1H),2.76(dd,J=17.6,7.6 Hz,1H),2.56(m,J=14.5,7.0,3.7 Hz,1H),2.27(d,J=4.9 Hz,1H),2.25-2.19(m,1H),2.18(dd,J=5.0,2.7 Hz,1H),2.09(s,3H),2.02-1.81(m,3H),1.73(t,J=13.2 Hz,1H);13C NMR(101 MHz,CDCl3)δ210.00,206.64,107.04,64.71,64.56,42.59,42.41,40.14,37.75,34.50,30.27.
化合物4的合成 称取3 g(14.1 mmol)化合物3加入到250 m L反应瓶中,加入100 m L的THF,强力搅拌,缓慢滴加2 M HCl(aq)40 m L.保持温度30℃反应6 h,TLC监控完全反应,向反应液中缓慢加入饱和Na HCO3溶液,调节p H至中性。CH2Cl2萃取,无水硫酸钠干燥,减压蒸馏得粗品1.9 g。将粗品加入到300 m L 的反应瓶中,加入甲醇200 m L强力搅拌,缓慢滴加2 M KOH(aq)20 m L。加热75℃回流3 h停止加热,待反应液冷却,加入柠檬酸调节PH ≈8,用50 m L CH2Cl2萃取3次,合并有机相,无水硫酸钠干燥,减压浓缩色谱柱分离(石油醚:乙酸乙酯1:2),得棕色粉末状固体1.5g。收率72%。
1H NMR(400 MHz,DMSO)δ5.48(s,1 H),2.75(dd,J=12.1,6.4 Hz,1 H),2.55-2.49(m,2 H),2.48-2.34(m,2 H),2.32-2.26(m,1 H),2.25(d,J=3.7 Hz,1 H),2.09-2.00(m,1 H),1.92(dd,J=17.4,4.3 Hz,1 H),1.45(dt,J=12.4,7.3 Hz,1 H);13CNMR(100 MHz,DMSO)δ177.62,40.61,40.40,40.19,39.98,39.77,39.56,39.35,38.19,28.13.
结果与讨论
本研究利用简单易得的原料1.3-环己二酮单乙二缩酮经过4步转换,以良好的收率合成双环[4.3.0]环戊烯酮,为构建天然产物分子中具有双环[4.3.0]结构的天然产物提供实用方法。
猜你喜欢
世界农药(2022年10期)2022-11-10
炼油技术与工程(2022年8期)2022-08-18
科学导报(2022年41期)2022-07-13
安徽农学通报(2022年8期)2022-05-06
能源化工(2021年2期)2021-12-30
昆明医科大学学报(2021年8期)2021-08-13
农药科学与管理(2021年2期)2021-03-16
读与写·下旬刊(2018年6期)2018-07-14
科学与财富(2017年17期)2017-06-16
家用汽车(2016年12期)2017-02-09
