
城市五种景观植物土壤微生物多样性研究①
2021-11-01 06:29魏岩冰曹舒阳钱宇杰
土壤 2021年4期
任 一,韩 畅,杨 慧,魏岩冰,曹舒阳,钱宇杰,唐 赟
城市五种景观植物土壤微生物多样性研究①
任 一1,2,韩 畅2,杨 慧2,魏岩冰3,曹舒阳3,钱宇杰3,唐 赟1*
(1 西南野生动植物资源保护教育部重点实验室,四川南充 637009;2 上海美吉生物医药科技有限公司,上海 201318;3 上海市川沙中学,上海 201299)
绿地为城市提供广泛的生态服务,绿地土壤微生物群落是其中的关键组成部分。为探究城市绿地土壤微生物多样性及其影响因素,通过16S rRNA基因的高通量测序对城市绿地5种常见景观植物土壤微生物进行分析,发现不同景观植物土壤微生物优势物种具有较高的一致性,以变形菌门、放线菌门、酸杆菌门、绿弯菌门、粘球菌门为优势门。但其α多样性和β多样性根据景观植物不同表现出明显相异性。另外,各土壤中古菌趋同性极高,几乎都属于氨氧化古菌。微生物相互作用关系主要受土壤含水量、盐离子浓度和pH等因子的影响。
城市微生物组;高通量测序;城市绿地;微生物多样性
近年来,我国人口城镇化进程的不断提速,城市化率从20世纪90年代的26.4% 持续上升到2019年的60.6%,预计到2035年,中国城镇人口比例将达到70% 以上。随着大量人口的涌入,打造更宜居的城市生态系统越来越受到政府和广大群众的重视。
随着大量的研究揭示微生物群落对人类疾病发生和维持人体健康平衡有重大影响,城市环境微生物群落(urban microbiome)对城市居民身心健康福祉的重要性逐渐受到大家的重视。城市环境微生物群落泛指城市各功能区块中的所有微生物群落,无论在这些功能区块的土壤、空气、水还是物体表面。大量的研究表明婴幼儿早期接触环境微生物的多寡与其哮喘的发生密切相关[1];经常进行户外活动的人比呆在家里的人拥有更多的微生物多样性,微生物多样性对人体免疫器官的发育、肠道营养物质的代谢至关重要[2];太多时间暴露在户外的人群又增加了获得感染性疾病的风险[3];还有一些研究表明城镇化水平的不同,其空气微生物和潜在致病微生物的组成存在差异,空气微生物组(25.6 ~ 124.4 cfu/m3)的多样性程度远远低于土壤微生物[4]。土壤微生物不仅是生态系统的维持者,还是大气环境微生物的主要来源之一,可通过人体的呼吸道和受损体表等暴露界面对人体健康产生影响,对这些生态系统中的土壤微生物群落的研究就显得尤为重要和急迫[5]。城市绿地如公园、小区绿化、道路绿化带等绿色基础设施是绿色城市生态系统中的重要组成部分,也是城市生态系统生物多样性的重要载体[6]。在城市绿化建设中人们通常更重视景观植物在改善空气质量、降低噪音、提升环境的整体观赏性、增强舒适度等方面发挥的作用[7],对城市绿地土壤微生物群落的研究长期以来一直被忽视,导致我们对其多样性及其影响因素的认识非常有限。
本文以上海市城市绿地常见景观植物文旦柚((Burm.) Merr.cv.Wentan)、凤尾竹((Lour.) Raeusch.ex Schult.'Fernleaf' R.A.Young)、樱花((Lindl.) G.Don ex London)、垂柳(L.)和香樟((L.) Presl)等土壤为研究对象,利用土壤理化因子测定和基于Illumina MiSeq平台的高通量16S rRNA基因测序技术,有效解决测序通量低和准确率低的缺陷[8]。本文研究了上述景观植物土壤微生物的丰度和群落结构以及土壤的理化性质对微生物群落的影响,以期增进人们对于城市绿地生态系统植物根系环境对微生物群落结构和功能塑造的理解,为绿色城市生态建设提供理论依据。
1 材料与方法
1.1 研究区概况
研究区位于上海市,地处长江入海口,太湖流域东缘,是我国南北海岸线中心点,长江携带的大量泥沙形成了上海市江海相沉积物的成土母质。该区属亚热带季风性气候,日照充足,温和湿润,雨量丰沛,四季分明,春秋两季较短,冬夏两季较长,年平均气温20 ℃,平均降水量1 512 mm,全年约70% 的降水量集中在5—9月。
1.2 供试材料
选取上海市川沙中学校园内不同地点的5种景观植物:文旦柚((Burm.) Merr.cv.Wentan)、凤尾竹((Lour.) Raeusch.ex Schult.'Fernleaf' R.A.Young)、樱花((Lindl.) G.Don ex London)、垂柳(L.)、香樟((L.) Presl),每种植物各选取3棵生长状态良好的植株,围绕各株植物设置3个采样点,采样时去除地表杂质,取深度15 ~ 20 cm根部附近土壤,将3个采样点土壤混合均匀后,封装于无菌聚乙烯自封袋中,置于干冰上运输至实验室。

表 1 土壤采样点信息
1.3 试验方法
1.3.1 土壤理化性质测定 土壤理化性质测定参照《土壤农化分析方法》[9]。土壤样品过20目筛(孔径0.9 mm),去除土壤中植物残留、碎石及其他杂物等。土壤含水量采用烘干法测定;土壤pH和电导率采用 pH/离子多功能测量仪(SevenMulti S40, Mettler Toledo, Switzerland)测定(土水比1︰5,︰);可溶性有机碳采用总有机碳/氮分析仪测定(非分散红外吸收法);采用流动分析(CFA和FIA)和光谱检测法测定水相中的可溶性总氮、硝态氮和铵态氮含量,可溶性有机氮(SON)=可溶性总氮–(硝态氮+铵态氮)。土壤有效磷测定采用碳酸氢钠–钼锑抗混合显色–分光光度计法。
1.3.2 土壤微生物总DNA提取 采用Fast DNA Spin kit for soil试剂盒(MP Biomedicals,美国)提取土壤总DNA样品。在无菌操作环境下,取0.5 g土壤样品,根据试剂盒操作说明提取土壤微生物总DNA,并使用Nanodrop ND-1000 紫外可见分光光度计(NanoDrop Technologies, Wilmington, DE, 美国)和1% 的凝胶电泳检测DNA浓度和质量,将DNA于–20 ℃ 保藏。
1.3.3 PCR扩增利用 “地球微生物组计划”(Earth Microbiome Project)官方推荐的用于土壤微生物16S rRNA基因通用引物515F (5’-GTGYCAGCMG CCGCGGTAA)和806R (5’-GGACTACNVGGGTW TCTAAT)(https://earthmicrobiome.org/protocols-and-standards/16s/),对土壤细菌和古菌的16S rRNA基因序列的可变区(V4)进行扩增。本次扩增使用的PCR仪型为ABI GeneAmp® 9700型。扩增体系总体积为20 µl。扩增体系具体描述如下:2 µl 2.5 mmol/L dNTPs,0.8 µl 引物(5 µmol/L),4 µl 5×FastPfu缓冲液,0.4 µl FastPfu 聚合酶, 10 ng DNA作为模板。PCR程序设定参数如下:95 ℃ 预变性3 min,设置27个循环,95 ℃ 变性30s,55 ℃ 退火30s, 72 ℃ 延伸30 s,72 ℃ 延伸 10 min。反应结束后,4 ℃ 保存。
1.3.4 Illumina MiSeq测序与数据分析 PCR产物采用2% 琼脂糖凝胶电泳检测,对目标PCR条带进行切胶,然后利用AxyPrep DNA 凝胶提取试剂盒(Axygen Biosciences, Union City, CA, USA)进行回收和纯化。利用QuantiFluor™-ST (Promega, USA) 定量检测PCR产物含量。本实验流程严格根据Illumina MiSeq平台 (Illumina,USA)标准流程,将目的扩增片段构建文库(PE 2×300),进行16S rRNA基因的高通量测序。原始高通量下机数据的质控和拼接,分别采用FLASH 1.2.700[10]和FASTP 0.19.600[11]软件。具体设置参数如下:①窗口长度为50 bp,当窗口内平均质量低于20时,从窗口开始截去后端碱基,最后去除质控后长度低于50 bp的reads;②去除含N碱基的reads;③去除引物错配属大于2的reads;④将overlap≥10 bp的paired-end序列进行拼接。OTU聚类与物种注释方法:①使用QIIME 1.9.10[12]对拼接后的序列进行去重复,得到unique序列;②根据97% 的相似度对unique序列进行OTU聚类[13](UPARSE 7.0.1090[14]);③通过将OTU代表序列与SILVA 132数据库进行比对(RDP Classifier 2.11[15]),得到每个OTU的物种注释信息;④基于注释信息,去除样本中的叶绿体和线粒体污染;⑤使用所有样本中的最小序列数进行抽平。数据统计与分析方法具体如下:①Alpha多样性指数间差异检验使用Student’s t-test进行差异显著性分析;②PcoA、db-RDA和PERMANOVA采用R vegan;③使用Kruskal-Walls秩和检验进行差异微生物分析;④采用Spearman指数来衡量微生物与土壤理化指标之间的相关性;⑤通过python包NetworkX 分析OTU互作网络。本研究利用美吉生物云平台(https://cloud.majorbio.com/)完成高通量测序数据分析任务。美吉生物云平台是本研究团队自主开发的生物信息学在线分析平台,平台整合了微生物多样性研究领域普遍认可的mothur1.30.1[16]( http:// www.mothur.org)、R Vegan(https://cran.r-project.org/ web/packages/vegan/index.html)、Circos-0.67-7 (http:// circos.ca/)等分析软件,目前支撑了国内外2 000多家科研单位的生物信息学分析。
2 结果
2.1 土壤理化性质与植物特性
5种景观植物分布于校园不同区域,其栽种土壤类型一致,均是含砂质壤土,校园景观植物无施肥管理。因此除个别样品外,土壤基础理化性质基本一致(表2)。土壤 pH范围8.28 ~ 8.65。土壤含水量132.3 ~ 230.6 g/kg,电导率(EC)范围115.00 ~ 147.60 μS/cm。然而土壤养分存在较大差异,可溶性有机碳(DOC) 40.00 ~ 128.95 mg/kg,可溶性有机氮(DON)4.48 ~ 11.94 mg/kg,可溶性总氮(TDN)7.51 ~ 18.14 mg/kg,硝态氮和亚硝态氮0.67 ~ 4.42 mg/kg,铵态氮0.46 ~ 2.76 mg/kg,有效磷3.97 ~ 29.42 mg/kg。文旦柚好高温不耐寒,在各种土壤中均可栽培;樱花树是我国华东地区常见的景观植物,根系较浅,不耐水湿;凤尾竹和樱花树喜弱酸性土壤;而垂柳和香樟是我国长江流域广泛种植的高大乔木,是城市中常见的景观植物,两种植物对土壤要求不高。
2.2 微生物多样性
MiSeq高通量测序共获得734 281 个16S rRNA基因序列,平均每个样本48 952个序列,序列平均长度320 bp。以测序数据量最少的样本为基准,对每个样品的16S rRNA基因序列进行抽平,最小数据量为30 333 条序列,然后根据抽平后的序列进行基于16S rRNA基因的微生物多样性和组成分析。
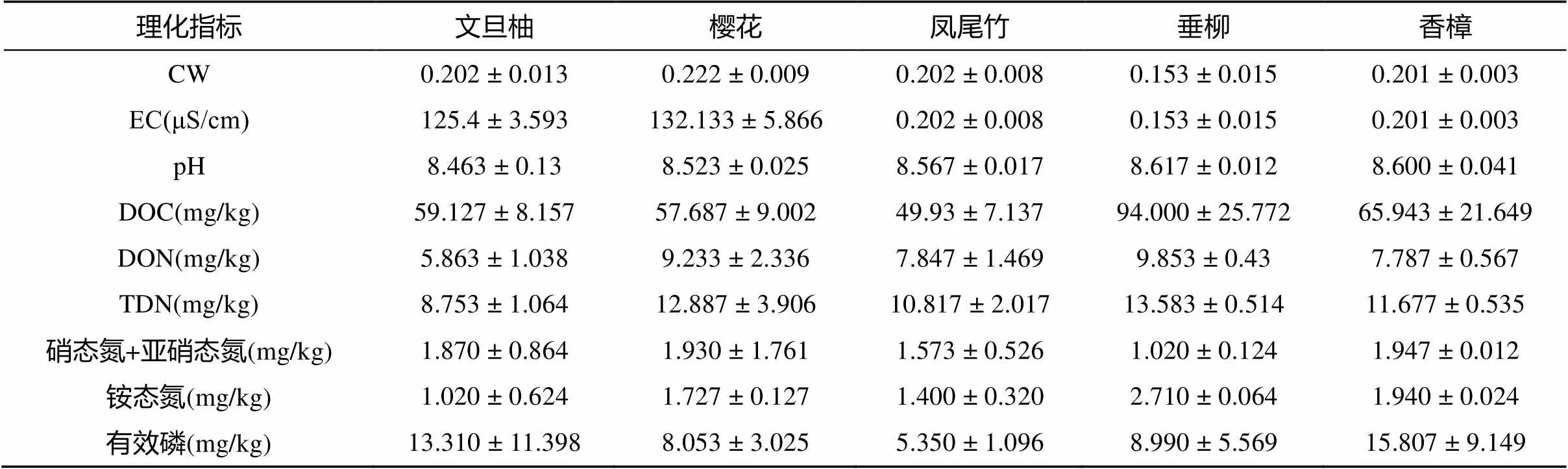
表 2 土壤理化性质
2.2.1 土壤微生物群落组成 本研究采用的高通量测序引物是“地球微生物组计划”(Earth Microbiome Project)官方推荐的能够同时覆盖细菌和古菌16S rRNA基因序列的通用引物[17]。5种景观植物附近土壤中细菌类群一致,都包含变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、粘球菌门(Myxococcota)、芽单胞菌门(Gemmatimonadetes)、浮霉菌门(Planctomycetes)、Methylomirabilota、疣微菌门(Verrucomicrobia)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、Latescibacterota、NB1-J等(图1A)。此外,奇古菌(Thaumarchaeota)在各个土壤中也有显著分布,占原核微生物总丰度的5.5% ~ 13.1%(图1A),占古菌丰度的98%。总体来看,变形菌、放线菌、酸杆菌、奇古菌和绿弯菌是各个土壤中排前5位的优势门,它们总相对丰度分别可占凤尾竹、樱花、香樟、垂柳、文旦柚土壤原核微生物的78%、76.5%、75.3%、82%、77.7%(图1A)。韦恩图分析也表明,5种景观植物土壤微生物组成保守性很高,其共有核心OTU为1 651个,占比OTUs总数的83.1%,表明校园绿化带土壤微生物主要类群有较高的同质性(图1B)。

图 1 门水平微生物群落组成(A)和OTU韦恩图(B)
微生物群落与理化因子相关性分析(图2)显示门水平总丰度前30的土壤微生物与土壤理化指标之间的相关性关系,其中放线菌(Actinobacteriota)与可溶性有机氮显著正相关(<0.05),绿弯菌(Chloroflexi)与可溶性有机氮显著负相关(<0.05)。Methylomirabilota与盐离子浓度和铵态氮呈显著负相关(<0.01)。浮霉菌(Planctomycetota)与土壤含水量显著负相关(<0.05)。厚壁菌门(Firmicutes)与有效磷为极显著正相关关系(<0.001)。
2.2.2 Alpha多样性 Sobs和Chao指数显示各植物土壤微生物群落的物种丰富度接近(图3A,3B),与OTU组成比较结果一致。在群落多样性方面,文旦柚的Simpson指数最高,樱花Simpson指数最低,文旦柚与凤尾竹、樱花、香樟的Simpson指数具有显著性差异(图3C)。
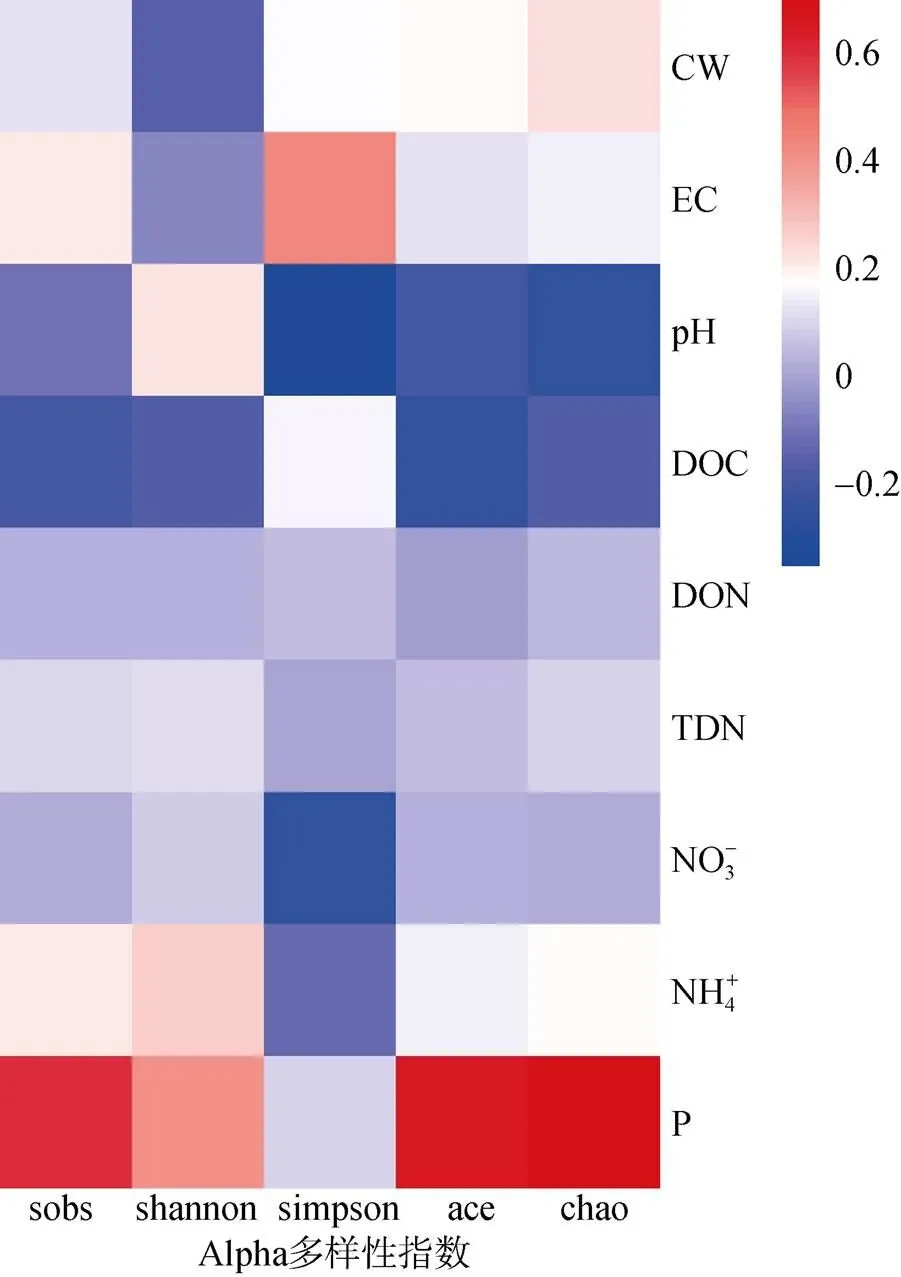
土壤微生物群落Alpha多样性与土壤理化因子相关性分析(图4)显示,土壤中的有效磷含量与Sobs、Shannon、Ace和Chao指数呈明显的正相关关系,与Simpson指数呈明显的负相关关系,表明土壤中的微生物多样性与磷元素正相关。土壤中的可溶性有机氮、可溶性全氮以及硝态氮与土壤微生物多样性表现出负相关关系。
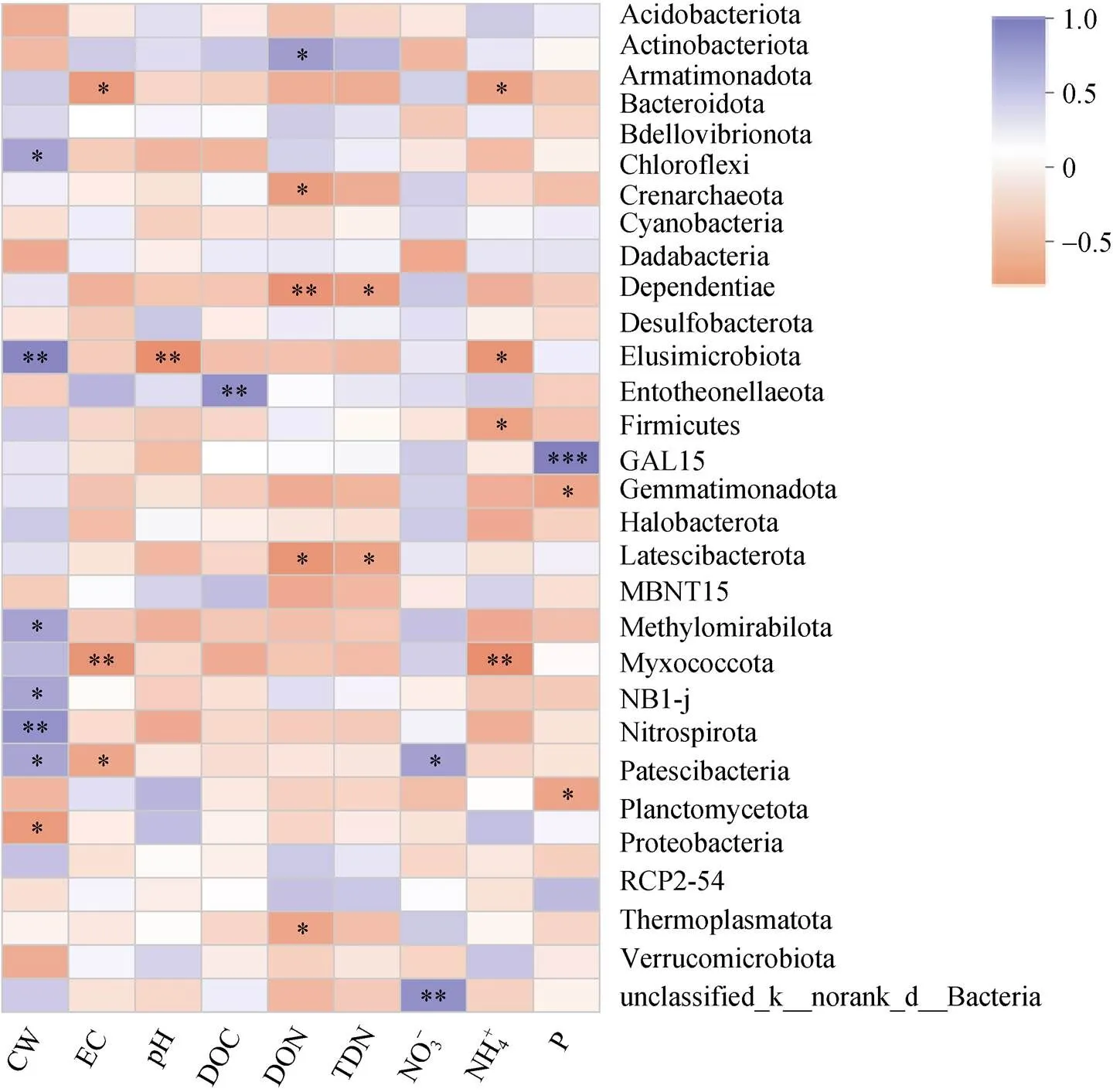
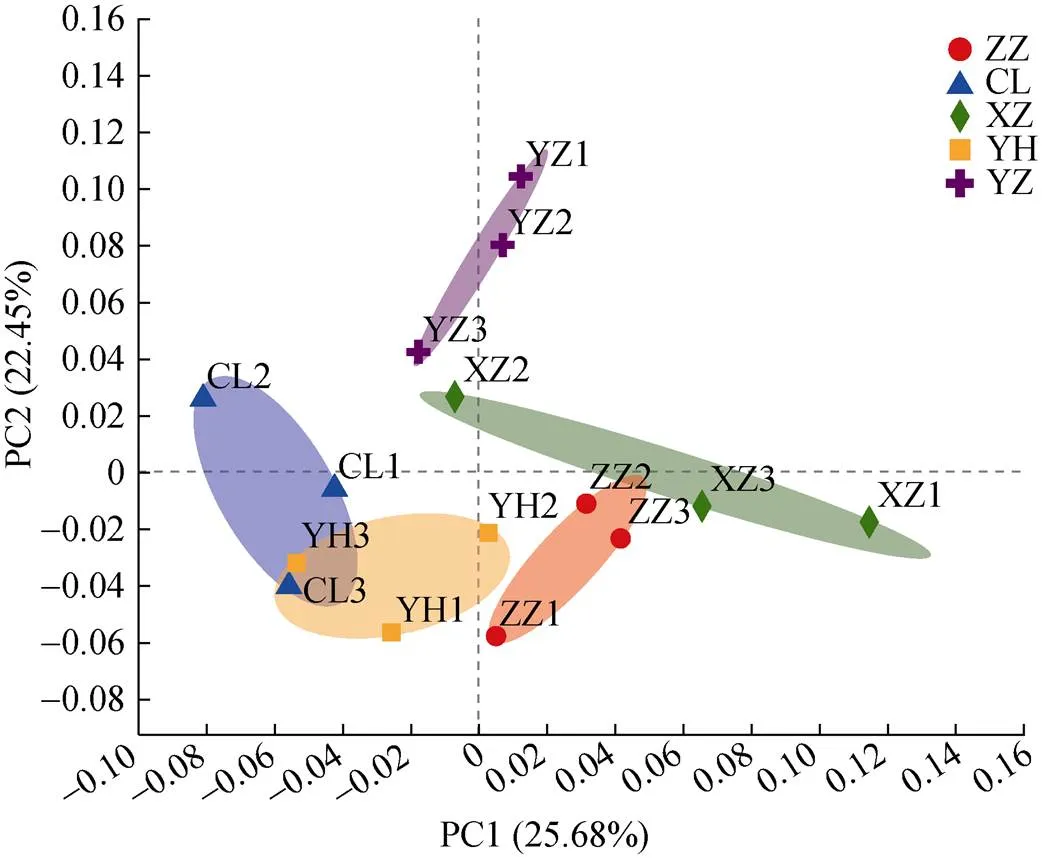
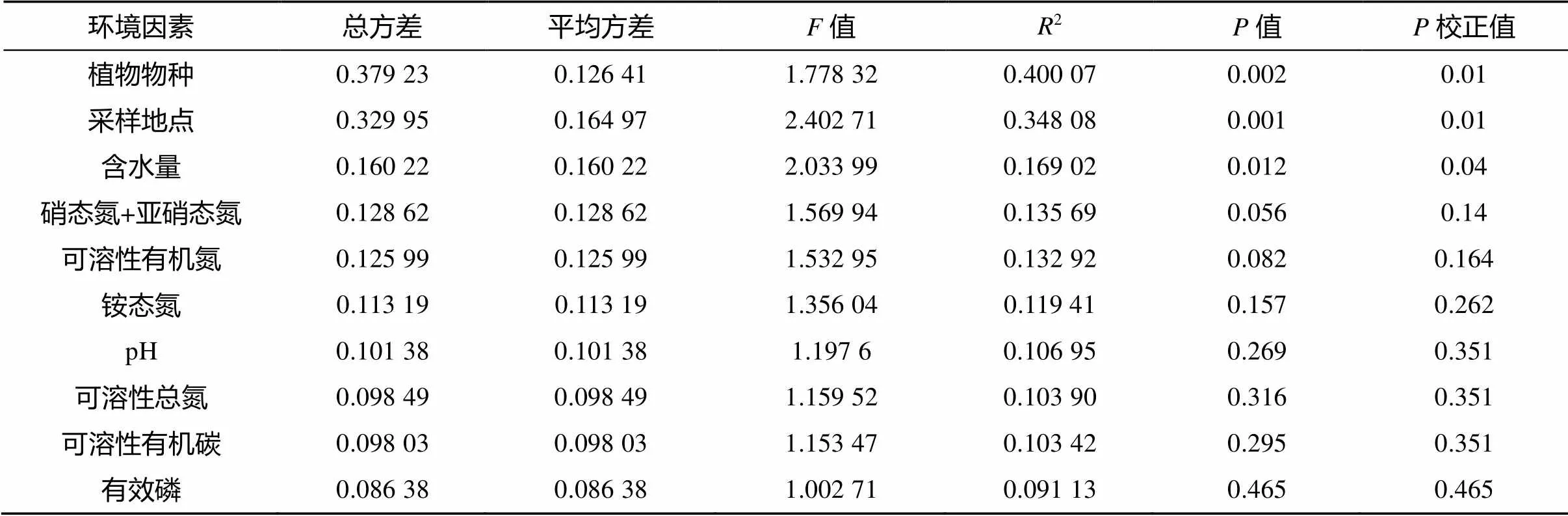
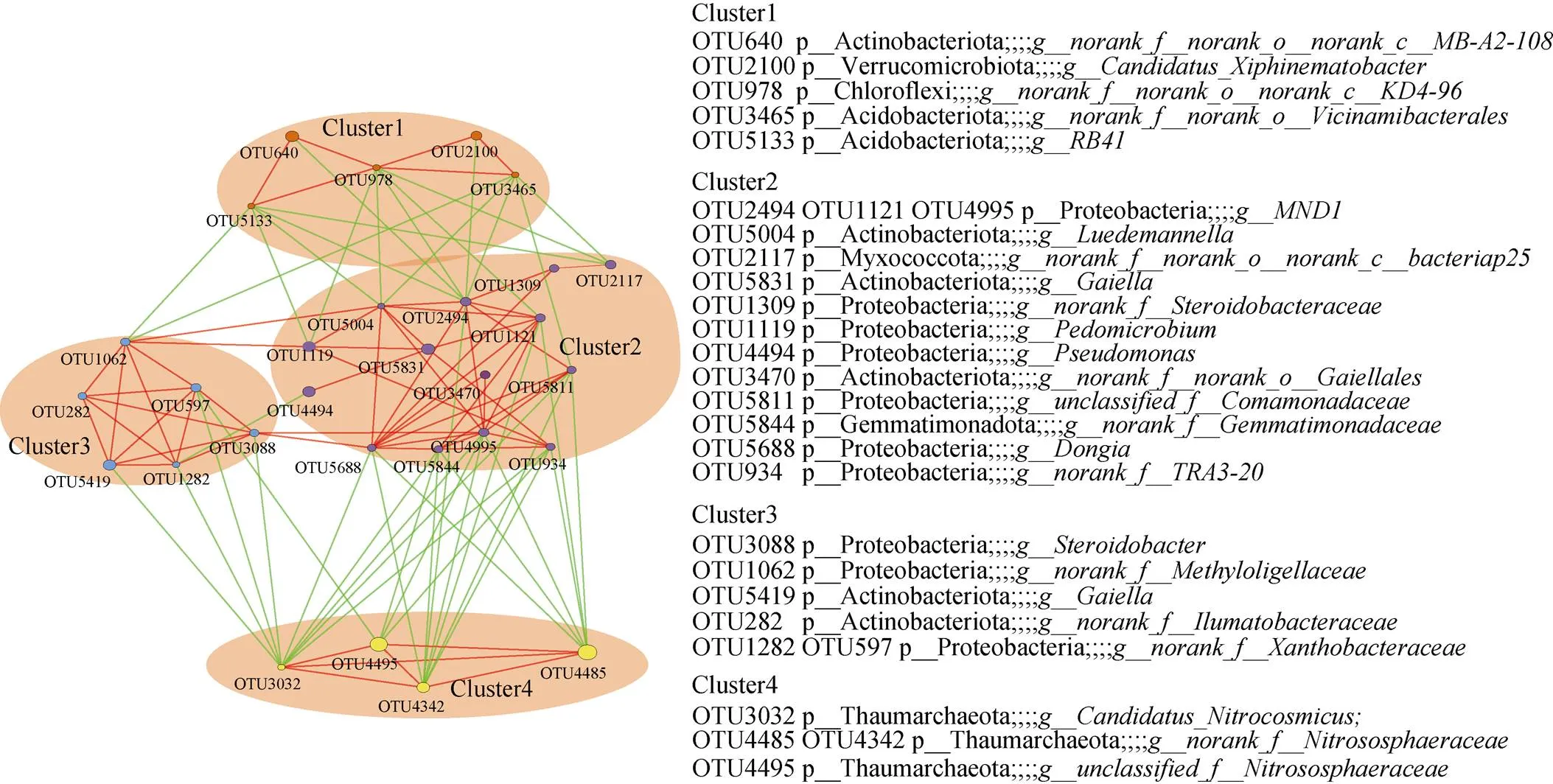
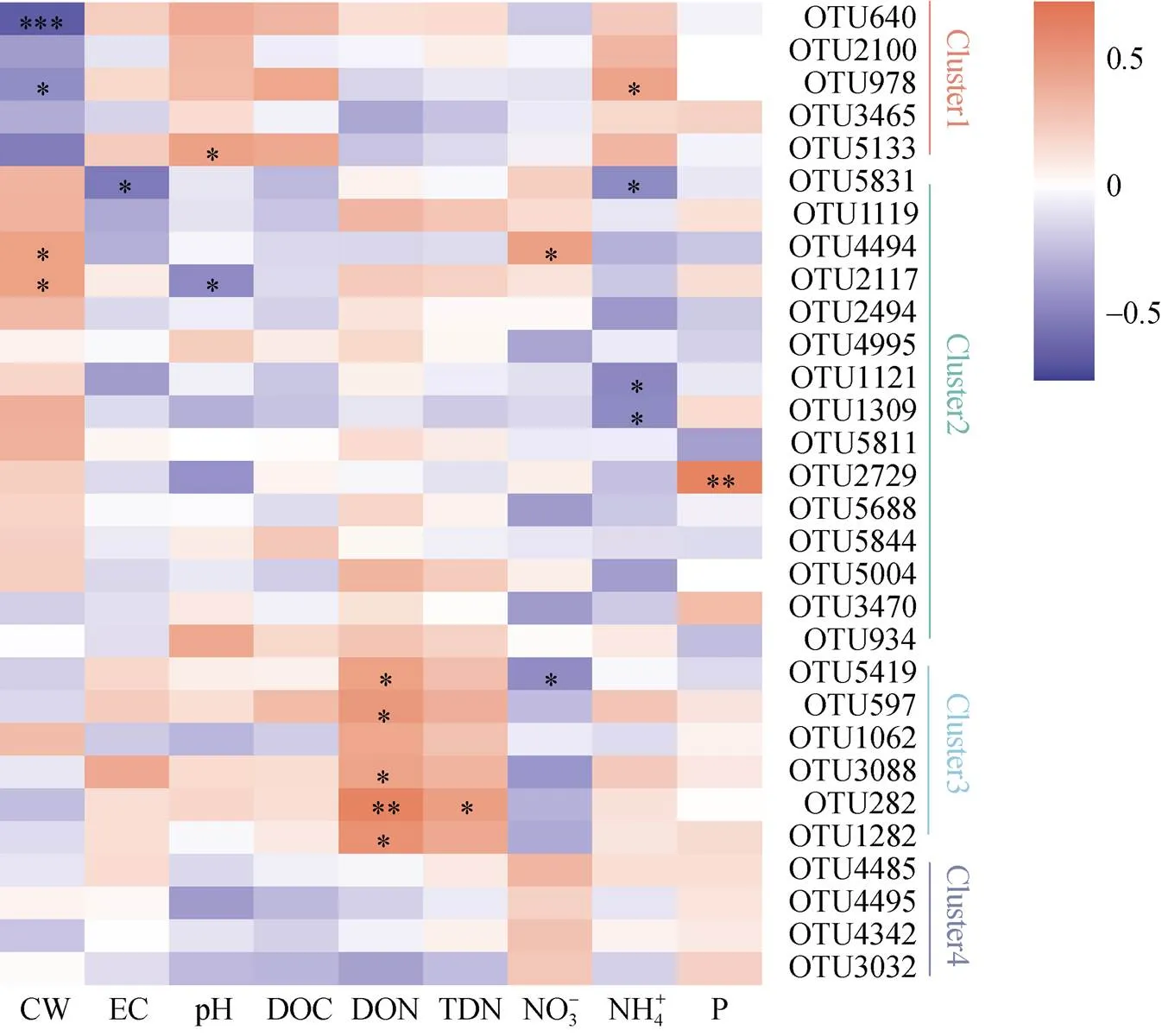
(*、**、***分别表示相关性达0.01 (图C中 *代表显著性差异(P<0.05)) 2.2.3 Beta多样性 Beta多样性用于表征不同微生态环境中的物种变化更替和物种组成差异。基于考虑物种丰度的Jaccard距离的PCoA(主坐标分析),前两个主成分将不同的植物土壤样本有效区分(图5),表明不同植物土壤样本中的细菌主要类群虽然有较高同质性,但总体微生物结构仍存在明显差别。同时我们发现取样地点也有一定的聚集效应,如位于河畔的3号樱花土壤样本和河畔的垂柳土壤样本距离接近,在菌群组成上相似度较其他樱花样本高(图5)。 PERMANOVA (permutational multivariate analysis of variance,置换多元方差分析)[18]表明不同的校园植物种类对于土壤菌群组成差异的解释度最高,其次为采样地点,再次为土壤中的含水量,其他土壤理化指标对于不同校园植物的土壤菌群的影响不具有显著性(表3)。 图4 土壤微生物Alpha多样性与环境因素相关性 2.2.4 OTU互作网络及其与理化指标相关性分析 本研究选择相对丰度较高的前30个OTU构建景观植物土壤微生物的互作网络(<0.05,>0.5)。网络显示土壤微生物群落中丰度最高的30个OTU主要分为4个簇(图6)。每个簇内部OTU丰度呈正相关,簇2、3分别与簇1、4呈负相关(图6)。簇1与土壤水含量呈负相关,其中OTU640(MB-A2-108)、OTU978(KD4- 96)和OTU5133与水含量呈显著负相关(图7);簇1与土壤氨态氮呈正相关,其核心节点OTU978与之显著相关(图7);簇2与土壤水含量呈正相关,其中OTU2117和OTU4494与之显著正相关(图7);簇2与土壤氨态氮呈负相关,其中OTU1121、OTU1309和OTU5831与土壤氨态氮呈显著负相关;簇3与NO– 3呈正相关,与土壤DON呈显著正相关(图7);簇4全部为奇古菌门氨氧化菌,与pH负相关,但并不显著(图7)。 图5 OTU水平PCoA 表3 不同因素对土壤菌群的显著性影响 城市绿化是城市生态环境的重要组成单元,是城市居民休闲、放松的重要场所之一,在这些户外场所中人们与微生物群落密切接触和互动,对人体健康产生重大影响。本研究采用高通量测序并结合土壤理化指标测定,对上海市常见景观植物的土壤微生物群落结构进行了探索。 研究发现景观植物根际附近土壤中微生物主要以变形菌、放线菌、酸杆菌、奇古菌、绿弯菌、芽单胞菌、浮霉菌为主要微生物,在总体原核微生物占比超过90%,与Manuel等人[19]的研究结论一致。城市景观植物的土壤微生物群落的物种丰富度非常接近,共同具有80% 以上核心OTUs,Sobs和Chao指数显示与其他野外生境相比[20],景观植物土壤微生物物种丰度偏低,这说明城市土壤微生物多样性存在缺失,且有同化趋势,这与农村与城市人群微生物多样性的差异结果是一致的[1]。古菌多样性的缺失最为明显,分析显示来自奇古菌的氨氧化古菌占土壤古菌总丰度的98% 以上,这与Dietrich等人[21-22]对其他大城市土壤微生物的研究一致。 (节点间红色连线表示正相关关系,绿色连线代表负相关关系) 图7 土壤微生物丰度(OTU水平)与环境因素相关性 本研究中,绿地景观植物生长所在的土壤类型相同,不同植物根部土壤中的微生物物种丰富度无显著差异,但不同植物根部土壤微生物多样性显示出一定差异,说明特定的群落结构是不同植物的根与土壤环境共同作用的结果。尽管不同景观植物土壤微生物主要类群比较一致,但其土壤微生物多样性仍具有明显差异,表明与自然生态系统一样[23],城市土壤微生物群落组成随着其宿主植物的变化而变化。这可能与微生物的宿主特异性、功能互补、根系结构等有关。植物根部土壤微环境的差异对土壤微生物群落结构差异产生一定影响。土壤有效磷含量对微生物与土壤微生物多样性表现出正相关关系,这与Gu 等人[24]发现在中国黑土中的微生物多样性与土壤中有效磷含量呈正相关关系一致,提示土壤中的有效磷可能是影响土壤微生物多样性的重要指标。Su等人[25]发现在水稻土壤中有效磷以及碳氮肥料的剂量增加,不改变微生物物种丰富性(species richness),而均一性(evenness)有所变化,土壤中有效磷剂量对土壤中的微生物多样性不产生显著性影响,但土壤中的菌群功能表现出显著性差异。 土壤微生物种群结构一般受土壤水含量、营养物质、pH等环境因子显著影响。本研究发现放线菌与土壤可溶性有机氮显著正相关(<0.01,=0.62),土壤可溶性有机氮来源于土壤微生物对于有机氮的分解矿化作用,而放线菌绝大多数为化能异养型细菌,可以分泌大量的胞外酶促进有机物如含氮聚合物的分解从而形成可溶性有机氮。绿弯菌是一类光能自养型细菌,有机氮促进化能异养型细菌的生长,从而对光能自养型细菌的生长产生抑制,所以绿弯菌与土壤可溶性有机氮呈显著负相关。Rokubacteria是近年来新发现的在土壤中广泛存在的一类参与硫循环、氮循环(硝酸盐还原)的重要微生物[26],由于硝酸盐还原酶NarB的活性受到氨态氮的负调控[27],所以表现为Rokubacteria含量与土壤氨态氮浓度呈负相关。 微生物群落互作网络反映了高丰度物种在土壤中的互作关系。丰度较高的前30个物种形成4个微生物簇,其中古菌中奇古菌门中的一些氨氧化菌独立成簇。不同簇之间物种呈负相关,簇内部物种呈正相关,而这些成簇的微生物表现出与土壤水含量、pH以及含氮量的显著相关,说明土壤水含量、pH,特别是土壤氮含量是城市土壤微生物互作网络的主要影响因素。 通过高通量测序对城市绿地5种常见景观植物的土壤微生物群落分析发现,不同景观植物土壤微生物优势物种具有较高的一致性,但微生物物种多样性(α多样性)和群落结构(β多样性)随着植物品种的不同仍表现出明显的差异。另外,在空间距离比较接近的不同景观植物,其土壤微生物群落结构也会表现出较强的一致性。互作网络分析发现,土壤优势微生物种群聚类成4个主要簇,簇内微生物之间以正相关为主,而簇间正负关联性主要受土壤含水量、盐离子浓度和pH等因子影响。此外,各土壤中古菌趋同性极高,几乎都属于氨氧化古菌。 致谢:感谢中国科学院南京土壤研究所土壤与环境分析测试中心在土壤理化性质检测中给予的帮助! [1] Ayeni F A, Biagi E, Rampelli S, et al.Infant and adult gut microbiome and metabolome in rural bassa and urban settlers from Nigeria[J].Cell Reports, 2018, 23(10): 3056–3067. [2] Clemente J C, Pehrsson E C, Blaser M J, et al.The microbiome of uncontacted Amerindians[J].Science Advances, 2015, 1(3): e1500183. [3] Hanski I, von Hertzen L, Fyhrquist N, et al.Environmental biodiversity, human microbiota, and allergy are interrelated[J].Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(21): 8334–8339. [4] Li H, Zhou X Y, Yang X R, et al.Spatial and seasonal variation of the airborne microbiome in a rapidly developing city of China[J].Science of the Total Environment, 2019, 665: 61–68. [5] Ortiz G, Yagüe G, Segovia M, et al.A study of air microbe levels in different areas of a hospital[J].Current Microbiology, 2009, 59(1): 53–58. [6] 胡靖, 李彦林, 陈国榕, 等.城市绿地类型对土壤线虫多样性的影响及生物指示值分析[J].土壤, 2020, 52(3): 510–517. [7] 赵林林.校园绿化景观的功能及植物配置分析[J].农村科学实验, 2018(1): 49. [8] 赵帆, 赵密珍, 王钰, 等.基于高通量测序研究草莓根际微生物群落结构和多样性[J].土壤, 2019, 51(1): 51–60. [9] 王希通, 李梦醒, 刘淑英.土壤农化分析方法[M].河北人民出版社, 1980. [10] Magoč T, Salzberg S L.FLASH: fast length adjustment of short reads to improve genome assemblies[J].Bioinformatics, 2011, 27(21): 2957–2963. [11] Chen S F, Zhou Y Q, Chen Y R, et al.Fastp: an ultra-fast all-in-one FASTQ preprocessor[J].Bioinformatics, 2018, 34(17): i884–i890. [12] Gregory Caporaso J, Kuczynski J, Stombaugh J, et al.QIIME allows analysis of high-throughput community sequencing data[J].Nature Methods, 2010, 7(5): 335–336. [13] 邓超超, 李玲玲, 谢军红, 等.耕作措施对陇中旱农区土壤细菌群落的影响[J].土壤学报, 2019, 56(1): 207–216. [14] Edgar R C.UPARSE: Highly accurate OTU sequences from microbial amplicon reads[J].Nature Methods, 2013, 10(10): 996–998. [15] Wang Q, Garrity G M, Tiedje J M, et al.Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J].Applied and Environmental Microbiology, 2007, 73(16): 5261–5267. [16] Schloss P D, Westcott S L, Ryabin T, et al.Introducing mothur: Open-source, platform-independent, community- supported software for describing and comparing microbial communities[J].Applied and Environmental Microbiology, 2009, 75(23): 7537–7541. [17] Gregory Caporaso J, Lauber C L, Walters W A, et al.Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms[J].The ISME Journal, 2012, 6(8): 1621–1624. [18] Kelly B J, Gross R, Bittinger K, et al.Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA[J].Bioinformatics, 2015, 31(15): 2461–2468. [19] Delgado-Baquerizo M, Oliverio A M, Brewer T E, et al.A global atlas of the dominant bacteria found in soil[J].Science, 2018, 359(6373): 320–325. [20] Huang Q, Wang J L, Wang C, et al.The 19-years inorganic fertilization increased bacterial diversity and altered bacterial community composition and potential functions in a paddy soil[J].Applied Soil Ecology, 2019, 144: 60–67. [21] Epp Schmidt D J, Pouyat R, Szlavecz K, et al.Urbanization erodes ectomycorrhizal fungal diversity and may cause microbial communities to converge[J].Nature Ecology & Evolution, 2017, 1: 0123. [22] Epp Schmidt D J, Kotze D J, Hornung E, et al.Metagenomics reveals bacterial and archaeal adaptation to urban land-use: N catabolism, methanogenesis, and nutrient acquisition[J].Frontiers in Microbiology, 2019, 10: 2330.DOI:10.3389/fmicb.2019.02330. [23] Hoch J M K, Rhodes M E, Shek K L, et al.Soil microbial assemblages are linked to plant community composition and contribute to ecosystem services on urban green roofs[J].Frontiers in Ecology and Evolution, 2019, 7: 198.DOI:10.3389/fevo.2019.00198. [24] Gu S Y, Guo X J, Cai Y T, et al.Residue management alters microbial diversity and activity without affecting their community composition in black soil, Northeast China[J].PeerJ, 2018, 6: e5754. [25] Su J Q, Ding L J, Xue K, et al.Long-term balanced fertilization increases the soil microbial functional diversity in a phosphorus-limited paddy soil[J].Molecular Ecology, 2015, 24(1): 136–150. [26] Anantharaman K, Hausmann B, Jungbluth S P, et al.Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle[J].The ISME Journal, 2018, 12(7): 1715–1728. [27] 王朱珺, 王尚, 刘洋荧, 等.宏基因组技术在氮循环功能微生物分子检测研究中的应用[J].生物技术通报, 2018, 34(1): 1–14. Study on Soil Microbial Diversity Under Five Urban Landscape Plants REN Yi1,2, HAN Chang2, YANG Hui2, WEI Yanbing3, CAO Shuyang3, QIAN Yujie3, TANG Yun1* (1 Key Laboratory of Southwest China Wildlife Resources Conservation (Ministry of Education), Nanchong, Sichuan 637009, China; 2 Shanghai Majorbio Biomedical Technology Co., Ltd., Shanghai 201318, China; 3 Shanghai Chuansha High School, Shanghai 201299, China) Greenspaces provide extensive ecosystem services for urban,while soil microbiome are key components of Urban landscape.In this study, the high-throughput sequencing of 16S rRNA genes was used to analyze soil microbes under five landscape plants in urban green space in order to study the diversity and influencing factors of soil microbes.It was found that the dominant species of soil microbes had high consistency under different urban landscape plants, the advantaged bacteria at phylum level were Proteobacteria, Actinobacteria, Acidobacteria, Chloroflexi, Myxococcota, but with variety in alpha and beta diversity indexes.Moreover, taxonomic convergence was found in archaeal community, which nearly all belonged to ammonia oxidizing archaea.The interactions between different microorganisms were regulated by soil moisture, salt ion concentration as well as pH. Urban microbiome; High-throughput sequencing; City greenspace; Microbial diversity S154.36 A 10.13758/j.cnki.tr.2021.04.011 任一, 韩畅, 杨慧, 等.城市五种景观植物土壤微生物多样性研究.土壤, 2021, 53(4): 746–754. 四川省科技厅应用基础研究项目(2020YJ0346)资助。 (tangyun_502@cwnu.edu.cn) 任一(1979—),男,山西翼城人,博士,工程师,主要从事微生物进化、微生物生态相关的研究工作。E-mail: yi.ren@majorbio.com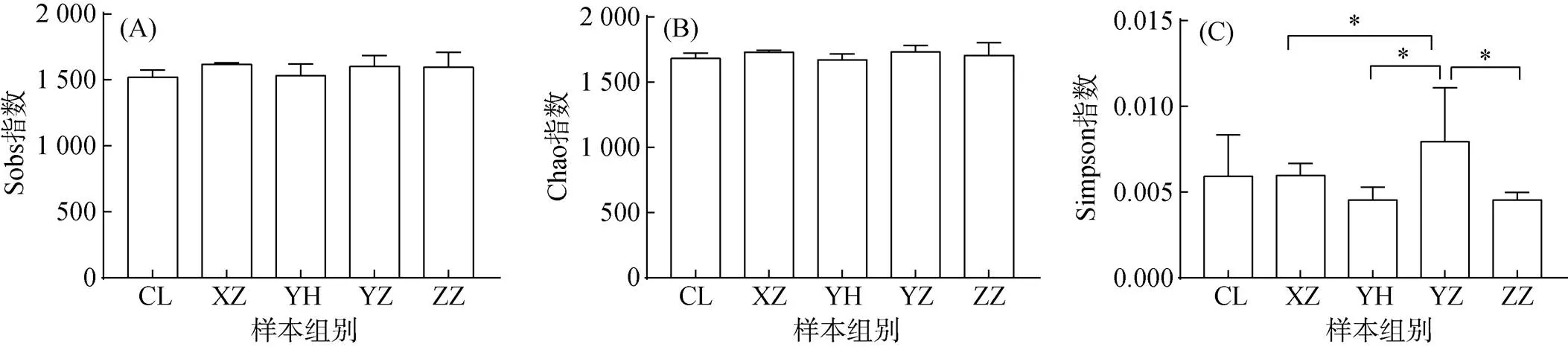
3 讨论
4 结论
猜你喜欢
当代水产(2022年8期)2022-09-20
卫星应用(2022年3期)2022-05-23
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年11期)2022-05-13
中国新通信(2022年4期)2022-04-23
昆明医科大学学报(2022年2期)2022-03-29
天津医科大学学报(2021年1期)2021-12-05
学校教育研究(2020年7期)2020-04-09
读写算(2018年6期)2018-08-22
考试周刊(2016年63期)2016-08-15
