
Fe2O3/石墨烯/埃洛石杂化气凝胶的电化学性能研究
2021-10-25 08:49余朝润王建宇李洪彦
湖南城市学院学报(自然科学版) 2021年5期
钟 勇,余朝润,李 棋,丁 炜,王建宇,李洪彦
(天津城建大学 材料科学与工程学院,天津 300384)
石墨烯气凝胶是由二维石墨烯片层三维搭接、堆叠而成的具有多孔结构的材料,其比表面积和孔隙率高,电化学性能好,在超级电容器领域中被广泛应用.人们在对石墨烯的研究中发现,三维互连的石墨烯气凝胶结构,使石墨烯具有多维电子传递途径,容易获得电解质和电极之间的最小运输距离[1-3].但无论是石墨烯气凝胶还是在石墨烯上负载金属氧化物制备的电极材料,电化学性能都不高[4-7].
埃洛石具有储量丰富、绿色无污染、比表面积大、强度高等优点,在催化剂载体等领域有着较为广泛的应用.使用埃洛石和金属硫化物、层状双氢氧化物复合,制备的材料比电容高、循环性能好.但这种材料中的金属硫化物、层状双氢氧化物容易发生团聚现象,大幅降低了电极材料的电化学性能[8-9].为提升电极材料的性能,可以采用不同类型的材料进行复合.科研人员将PEDOT负载在HNTs上,合成氧化石墨烯,制备埃洛石-石墨烯杂化气凝胶(HGO).HGO具有优异电化学性能,储能性能好,循环稳定性高[10-13].为了进一步提高材料的电化学性能,可在埃洛石-石墨烯杂化气凝胶中加入Fe2O3.在电极材料中,Fe2O3因为比电容高、成本低且绿色环保被广泛运用.在充放电过程中,Fe2O3的体积变化导致自身粉末化,与电解液之间形成SiC膜,阻碍了体系中的离子交换,这降低了电极材料的电化学性能和循环性能.通过Fe2O3和HGO的复合,HGO大的比表面积为Fe2O3的体积形变提供了缓冲空间,提高了电极材料的性能[14-16].
基于此,本研究采用改进后的Hummers法合成GO,并在GO上负载Fe2O3,同时在HNTs上负载PEDOT,利用具有电化学活性的PEDOT连接HNTs和负载Fe2O3的GO,制备Fe2O3/石墨烯/埃洛石杂化气凝胶,并通过多种手段对其进行表征分析,深入研究其电化学性能.
1 实验部分
1.1 实验原料及试剂
原料与试剂:HNTs;石墨;盐酸(HCl),过氧化氢(H2O2)为化学纯试剂;浓硫酸(H2SO4),硝酸钠(NaNO3),高锰酸钾(KMnO4),无水乙醇(C2H5OH),环己酮((CH2)5CO),3,4-乙烯二氧噻吩(EDOT),过硫酸铵((NH4)2S2O8),六水合三氯化铁(FeCl3·6H2O),过氧化二苯甲酰(BPO)等,均为分析纯试剂;去离子水.
1.2 仪器设备
仪器设备:真空冷冻干燥设备(200 mL,南通市华安超临界萃取有限公司);真空气氛节能箱式电炉(SX-G03163Q,天津中环实验电炉有限公司);场发射扫描电子显微镜(SEM,HELIOS NanoLab 600i,美国FEI公司);傅里叶变换红外光谱仪(FTIR,Perkin-Elmer-2000,美国PERKINELMER公司);X射线光电子能谱仪(XPS,Escalab 250 X,美国Thermo Scientific Ltd.公司);比表面积/孔径分析仪(Quabrasorb SI-3MP,USA);CHI 660电化学工作站(中国上海辰华有限公司).
1.3 实验步骤
1.3.1 氧化石墨烯片负载金属氧化物
使用改进后的Hummers法合成氧化石墨烯(GO):将H2SO4(96 mL)、石墨粉(2 g)、NaNO3(2 g)加入三颈烧瓶中,在冰水中磁力搅拌45 min;加入KMnO4(15 g),依次在0和35 ℃下搅拌2 h;将所得混合物用去离子水(100 mL)稀释,升温至98 ℃,磁力搅拌2 h;向悬浮液中加入去离子水(200 mL)和30%过氧化氢(25 mL),混合均匀;在混合溶液中加入5%HCl溶液以除去金属离子,并用去离子水洗涤混合物直至上清液显示中性;将少量悬浮液放入真空干燥箱中,在80 ℃下干燥24 h,经离心分离获得1 mg/mL的GO溶液,并计算悬浮液中GO的浓度.
取2 g FeCl3·6H2O溶于10 mL的蒸馏水中,准备50 mL沸水,将FeCl3·6H2O溶液缓慢滴入,整个过程持续5 min,待完成后煮沸1 min,放置冷却至室温备用.
取500 mL 0.5 mg/mL GO分散液,在磁力搅拌下滴加50 mL FeCl3·6H2O胶体,继续磁力搅拌2 h;将所得悬浮液离心分离获取沉淀物,在N2氛围和200 ℃下煅烧4 h,可制得负载了αFe2O3的GO片,记为GO-αFe2O3[17-18].
1.3.2 埃洛石纳米管负载聚3,4-乙烯二氧噻吩
将0.25 mg的HNTs加至40 mL水中,超声分散30 min,滴入220 μL 3,4-乙烯二氧噻吩,磁力搅拌10 min,加入5 mmol过硫酸铵,持续搅拌24 h;配制1.33 mL浓度为8 mol·L-1的NaOH水溶液,加入上述分散液,在90 ℃下搅拌5 h,当反应完成后离心分离获得沉淀物,经干燥获得负载了PEDOT的HNTs,记为HNTs-P[19-20].
1.3.3 Fe2O3/石墨烯/埃洛石杂化气凝胶
将HNTs-P和GO-αFe2O3分别分散于环己酮中,制得2种分散液;均匀混合2种分散液,加入BPO(0.1 g),搅拌10 min,并超声分散2 h;将所得分散液移入四颈烧瓶中,在N2氛围和75 ℃下预聚合1.5 h;依次在50,80,90和100 ℃的油浴中聚合1.5,2,2和2 h;用乙醇置换环己酮,经冷冻干燥处理,获得Fe2O3/石墨烯/埃洛石杂化气凝胶,记为HGO-αFe2O3[21].
1.3.4 对照组样品
将改良Hummers方法合成的GO溶液密封在特氟隆高压釜中,在180 ℃下放置12 h;将高压釜自然冷却至室温,获得水凝胶;将所得水凝胶用乙醇进行溶液置换,经超临界干燥、老化,制得石墨烯气凝胶,记为GA.
将HNTs-P与未负载Fe2O3的GO分别分散于环己酮中,按1.3.3相同的步骤制备不负载Fe2O3的HGO.
1.3.5 电化学实验
将所制HGO-αFe2O3复合材料作为活性物质与导电剂(乙炔黑)和黏结剂(聚四氟乙烯)混合,磁力搅拌24 h,形成浆料;将该浆料均匀涂抹在1 cm×1 cm的泡沫镍上,在60 ℃下干燥,制得超级电容器的工作电极.三电极体系中的电解质为1 mol·L-1的KOH溶液,铂片作为辅助电极,饱和甘汞作为参比电极.
2 实验结果与表征
2.1 化学组成及结构分析
2.1.1 傅里叶变换红外光谱分析
HNTs,GO,GO-αFe2O3,HGO和HGO-αFe2O3的FTIR谱如图1所示.
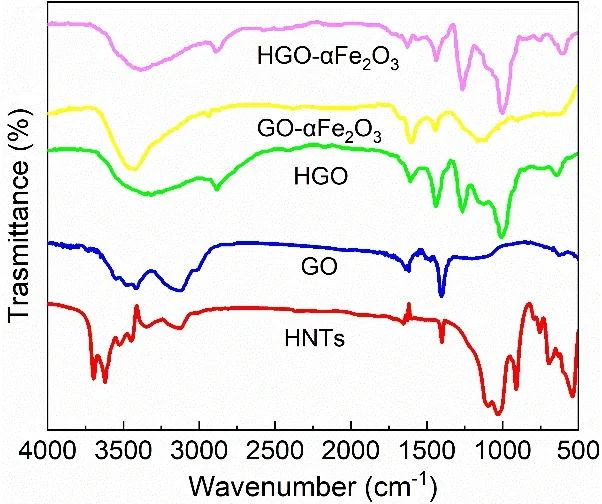
图1 HNTs,GO,GO-αFe2O3,HGO和HGO-αFe2O3的FTIR谱
由图1可知,在HNTs曲线中,531 cm-1附近是Al-O-Si键的特征峰;697,753和1 034 cm-1附近是不同表面的Si-O键的特征峰.HNTs内表面的O-H变形振动在911 cm-1处产生吸收峰,吸附水的O-H键在1 630 cm-1处产生吸收峰,HNTs内外表面的Al-OH键分别在3 696和3 625 cm-1处产生吸收峰[22].在GO曲线中,芳烃上的O-H键在3 224 cm-1处产生宽峰,1 627和1 408 cm-1附近的峰是羧基中的C-O键和环氧基团中的C-O-C键产生的.GO-αFe2O3曲线在553 cm-1附近出现了Fe-O键的吸收峰,在3 224 cm-1附近芳烃上的O-H键的峰强度也有所变化,这表明含氧基团被消耗.同时还发现,1 408 cm-1附近C-O-C键的吸收峰发生了一定的偏移,这说明GO和Fe2O3的结合与石墨烯的芳烃和环氧基团有关,且形成了Fe-O-C键.
在HGO的曲线中,Si-O-C振动产生了1 051和1 298 cm-1处的吸收峰.这是因为硅烷改性剂KH570中的Si(OCH3)3水解产生了Si-OH,Si-OH与GO表面的烷氧基作用,形成了Si-O-C键.这也说明在形成气凝胶前对GO改性成功.在HGO的曲线中,其组份的大部分特征峰都能呈现,如HNTs曲线的531 cm-1处Al-O-Si键和1 034 cm-1处Si-O键的特征峰.此外,HGO-αFe2O3与HGO曲线大致相同,这说明αFe2O3的加入没有对HGO产生太大的影响.
2.1.2 X光电子能谱分析
GO,HNTs,αFe2O3,HGO-αFe2O3,HGO和GA的XPS曲线以及GA和HGO的C1s谱如图2(a)~图2(c)所示.
由图2(a)可知,HNTs的XPS曲线在结合能为73,112和157 eV处出现Si2p和Si1p峰;αFe2O3和HGO-αFe2O3曲线均出现Fe2p峰;GO和GA曲线均出现C1s和O1s峰.这说明GO和GA仅由C和O组成.此外,HGO-αFe2O3曲线同时出现了C1s,O1s和Fe2p峰,这确定了其主要元素的存在.

图2 样品X光电子能谱
图2(b)中284.5 eV处的峰,说明GA存在C-C/C=C结构;286.5 eV处的峰,说明GA存在C-O结构;287.8 eV处的峰,说明GA存在C=O结构;289.1 eV处的峰,说明GA存在O-C=O结构.由于在GO至GA的合成过程中,GO被还原,其中的大部分含氧官能团被去除,所以可以观察到C-C/C=C的峰强度要远高于别的基团.
图2(c)的HGO的C1s分峰中含氧官能团明显变多,这可能是因为PEDOT中存在C-O,也可能是因为PEDOT具有还原性,在还原GO的同时会在GO上原位聚合生长,影响了少量GO的还原.这表明,PEDOT在HGO中的存在且在交联时起到了作用.
2.1.3 微观形貌分析
HGO-αFe2O3三维网络结构如图3所示.

图3 HGO-αFe2O3的SEM图
在图3中,明场为气凝胶3D骨架,暗场为气凝胶孔隙.由图3可知,HGO-αFe2O3气凝胶具有无定形和疏松的特点,GO与HNTs相互穿插搭建成气凝胶3D网络,大量的孔隙存在于其中,且由于GO负载了一定量的Fe2O3,GO片还呈现了一定厚度.实际上,包覆了PEDOT的HNTs与GO以多种方式相互连接,部分HNTs-P均匀分布在GO表面,在相邻的2片GO中也有HNTs-P的连接,并存在HNTs-P间相互连接的情况,由此才形成了HNTs-GO的杂化三维网络结构.在所形成的有机-无机杂化气凝胶中,HNTs与GO在HGO中均匀分布,并没有产生团聚现象,这是因为HNTs表面存在PEDOT,使PEDOT与GO之间存在各种化学键的连接与限制.
2.1.4 XRD测试分析
αFe2O3和HGO-αFe2O3的XRD分析图谱如图4所示.在图4展示的αFe2O3曲线中,与标准卡(JCPDS编号:33-0664)对比,各衍射峰的位置与(012),(104),(110),(214),(300),(119)和(220)面相符.对比HGO-αFe2O3和αFe2O3的XRD曲线,发现除了石墨烯的(202)和(002)晶面外,HGOαFe2O3存在和αFe2O3一样的衍射峰,这说明在HGO-αFe2O3中Fe以αFe2O3晶体的形式存在.

图4 αFe2O3和HGO-αFe2O3的XRD图
2.1.5 比表面积与孔径分析
GA,HGO和HGO-αFe2O3的N2吸附-脱附曲线及孔径分布如图5所示.

图5 GA,HGO和HGO-αFe2O3 N2吸附-脱附曲线及孔径分布曲线
由图5(a)可知,当相对压力较小时,N2吸收量较低,曲线变化平缓;当相对压力增加时,曲线呈明显的上升趋势;在P/P0=0.5~0.8处,各样品曲线均出现滞回环.图5(a)为带有H3滞回环的Ⅳ型曲线,此类曲线说明各样品中含有介孔结构,而滞后现象的发生是因毛细缩合排空和填充中孔所致.从图5(a)还可知,HGO比表面积大于GA,这归因于HNTs的加入连接了相邻GO,增加了HGO比表面积.但在HGO-αFe2O3中,Fe2O3并没有增加HGO的比表面积,反而使比表面积减少,这可能是因为夹杂在GO片层中的Fe2O3增加了HGO-αFe2O3的重量,导致HGO-αFe2O3的比表面积相对HGO减少了.
从图5(b)可以看出,HNTs的引入使得HGO相较GA而言,多出了不少微孔和介孔,这也是HGO相较GA其比表面积增大的原因,而在Fe2O3加入后,小孔径在气凝胶中明显减少.
2.2 电化学性能分析
对GA,HGO和HGO-αFe2O3进行电化学测试,分析其电化学性能.GA,HGO和HGOαFe2O3的循环伏安曲线(CV)和恒电流充放电曲线(GCD)如图6所示.
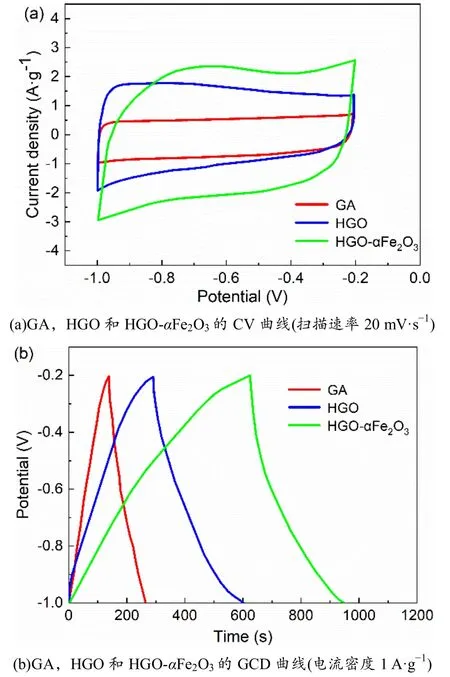
图6 样品电化学性能
图6(a)为在KOH水溶液中,扫速20 mV·s-1,电压窗口为-1~-0.2 V时的CV曲线.从图6(a)中可看出,GA的CV曲线十分规则且接近矩形,这是因为在GA的电化学测试过程中没有发生氧化还原反应,其电容器行为是双层电容.HGO的CV曲线没有GA的规则,这是因为在HGO中存在着少量的PEDOT,它会在电化学测试中发生还原反应,由原来的电中性还原为带正电荷,其电容器行为是双层电容和法拉第赝电容.PEDOT上的五元环和GO上的六元环产生π-π相互作用,大π键中的电子更具电化学活性,容易在2个π间移动,使得电流更容易通过包裹了PEDOT的HNTs到达GO,因此HGO的电化学性能高于GA.在HGO-αFe2O3的CV曲线中出现了明显的氧化峰,这是因为大量引入了Fe2O3,在充放电时Fe2O3发生了氧化还原反应:

在HGO和HGO-αFe2O3中,HGO-αFe2O3的法拉第赝电容占比更大,其提供了大量电容.CV曲线面积大小反映了被测样品的比电容大小,从图6(a)中CV曲线的面积可得知,三者的比电容大小关系为GA<HGO<HGO-αFe2O3.
图6(b)展示了在-1~-0.2 V的电位范围内,以1 A·g-1的电流密度对GA,HGO和HGO-αFe2O3进行恒电流充放电测试的结果.根据公式(2)可以计算所测样品的比电容.

其中,C(F·g-1)为比电容;I(A)为放电电流;Δt(s)为总放电时间;m(g)为电极材料质量;ΔV(V)为放电过程中的电位范围.
由图6(b)可知,GA,HGO和HGO-αFe2O3的比电容分别为156.6,357.9和449.5 F·g-1;GA和HGO的GCD曲线呈现较为标准的三角形,这说明GA和HGO以双层电容为主;HGO-αFe2O3的GCD曲线呈现一定的弧度,这说明在HGO-αFe2O3中包含了法拉第赝电容,该结果和CV曲线的分析一致.
为了进一步分析HGO-αFe2O3的电化学性能,还进行了其他电化学测试.
在KOH水溶液中,分别以10,50,100和200 mV·s-1扫描速率对HGO-αFe2O3进行循环伏安测试,结果如图7(a)所示.从图7(a)中可以看出,扫描速率越快,峰电流值就越大,氧化还原越明显,比电容中的赝电容增长大于双层电容增长,这是因为αFe2O3具备良好的电化学活性.由图7(a)还可以看出,100 mV·s-1扫描速率下的CV曲线和200 mV·s-1扫描速率下的CV曲线,前者存在更为狭窄的氧化还原峰,这可能是由于较高的扫描速率使得溶液中离子的运动速率过快,未能完全到达GO片层中的Fe2O3上.

图7 HGO-αFe2O3的其它电化学性能
进一步实验探究HGO-αFe2O3中的电化学机理,在-1~-0.2 V的电位范围内,以50 mV·s-1的扫描速率对3个样品进行循环伏安法测试,得到不同GO∶αFe2O3(质量比)的循环伏安曲线,如图7(b)所示.其中,HGO-αFe2O35,HGO-αFe2O310和HGO-αFe2O315对应的GO∶αFe2O3质量比分别为1∶5,1∶10和1∶15.由图7(b)可知,随着αFe2O3含量的增加,相应的CV曲线并没有均匀排布,这是因为αFe2O3不是直接负载在HGO中,而是存在于GO片层间.这种存在方式不仅会直接影响氧化还原反应的物质的量,还会改变GO片层间距,直接影响GO产生的双层电容大小,增加离子接触αFe2O3的难度.
图7(c)为3种不同αFe2O3含量的样品在不同电流密度下的比电容.不难发现,随着αFe2O3量的增加,HGO-αFe2O3整体比电容增加;随着电流密度增大,比电容减少.当扫描速率增加至4 A·g-1时,离子能够充分接触到αFe2O3,此时HGO-αFe2O315的比电容大于其他2种样品.
图7(d)为不同αFe2O3含量的HGO-αFe2O3循环测试结果.从图7(d)可知,当循环使用次数较少时,比电容会随循环次数的增加而增加.这是因为随着循环实验的进行,离子多次通过GO到达αFe2O3,使GO越来越松散,离子更容易接触到αFe2O3,使更多的αFe2O3能够参与反应.随着循环次数的继续增加,HGO-αFe2O3的比电容达到最高,随后会开始逐渐减小.这是因为随着循环实验的进行,αFe2O3的体积发生了变化,同时αFe2O3自身开始粉末化,与电解液之间形成SiC膜(粉末化程度越严重,SiC膜越厚),阻碍了体系中的离子交换.此外,因为αFe2O3是夹杂在GO片层之中的,所以αFe2O3的体积变化会对GO结构产生一定的破坏.经过1 000次循环实验,其比电容下降了6%~7%,该结果远优于以往报道中的GO-αFe2O3复合材料,这可能是因为HGO中刚性HNTs所组成的无机骨架在一定程度上缓解了GO-αFe2O3的体积变化.
3 结论
1)在HNTs上原位生长的PEDOT能够连接GO表面,形成GO/HNTs的双网络结构气凝胶.
2)HNTs的引入使得HGO比GA多出了不少微孔和介孔,对其比表面积的增大效果较为显著,在加入Fe2O3后,小孔径在气凝胶中明显减少.
3)HGO-αFe2O3比GA和HGO具有更高的比电容,这是因为材料中Fe2O3的存在提供了法拉第赝电容.
4)HGO-αFe2O3的循环性能得到很大提高,这是因为刚性HNTs所组成的无机骨架在一定程度上缓解了GO-αFe2O3的体积变化,对SiC膜的形成产生了影响.
猜你喜欢
小学生学习指导(高年级)(2022年3期)2022-03-29
防爆电机(2021年3期)2021-07-21
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
小学生学习指导(高年级)(2019年4期)2019-11-27
表面工程与再制造(2019年6期)2019-08-24
电子制作(2018年19期)2018-11-14
电子制作(2018年19期)2018-11-14
广东教育·高中(2018年12期)2018-02-13
小学生导刊(高年级)(2017年2期)2017-06-10
