
Immunotherapy for cold tumors
2021-10-13 07:32SUShengchenHyungLaeKim
中国临床新医学 2021年9期
SU Sheng-chen, Hyung Lae Kim
[Abstract] Immunotherapy has joined surgery, radiation, and traditional chemotherapy to become the fourth pillar in cancer treatment. The concept of using one′s own immune system to treat cancer has inspired numerous therapies. The class of immunotherapies that has proven most effective is the checkpoint inhibitors(CPIs), which work by blocking immunosuppressive mechanisms. Prime examples of CPIs in clinical use are based on Nobel Prize winning discovery of PD-1/L1 and CTLA-4. The clinical responses to immunotherapies vary, and this is in part due to differences in the tumor microenvironment. Tumors that are poorly infiltrated by lymphocytes are immunologically inert and are referred to as “cold tumors” and tend to respond poorly to CPIs. We review current immunotherapies and examples of efforts to increase lymphocyte infiltration in cold tumors to improve outcomes of CPIs.
[Key words] Cancer immunology; Immunotherapy; Tumor infiltrating lymphocyte
1 Common cancer immunotherapies
The past few decades have seen tremendous development in immunotherapies, which can boost or modify the immune system to find and attack the cancer cells[1]. Immunotherapies can target every step of the anti-tumor immune cycle: release of cancer cell antigens, antigen presentation, priming and activation of T cells, trafficking of T cell to tumors, infiltrating of T cells into tumors, recognition and killing of cancer cells(Figure 1). Common immunotherapies include monoclonal antibodies targeting tumor-intrinsic pathways and tumor-extrinsic factors, cytokines, cancer vaccines, adoptive transfer of ex-vivo-activated T cells and natural killers cells[2]. In this part of the review, we will briefly discuss the current status of important cancer immunotherapies.

Figure 1 The anti-tumor immune cycle and common immunotherapies. Adapted from Chen and Mellman[3]
2 Antitumor monoclonal antibodies
Most of the monoclonal antibodies(MABs) are specially engineered, humanized, or chimeric antibodies targeting tumor antigen or other proteins that promote tumor progression. Many MABs have been approved recently by the FDA, and are used to target and kill cancer cells[4]. For example, overexpression of HER-2 on tumor cell is associated with aggressive behavior in breast cancer. Trastuzumab(Herceptin), a humanized monoclonal antibody against the extracellular domain of HER2, has been shown to benefit patients with HER2-positive metastatic breast cancer. One year of treatment with trastuzumab after adjuvant chemotherapy significantly improves disease-free survival among women with HER2-positive breast cancer[5].
Other MABs work by blocking antigens on other cells that facilitate tumor growth. Bevacizumab(Avastin) targets the vascular endothelial growth factor(VEGF) A, which is a potent proangiogenic growth factor that stimulates the proliferation, migration, and survival of endothelial cells[6]. Blocking VEGF inhibits angiogenesis at tumor site, thus decreases oxygen flow and nutrient transport to the tumor. Bevacizumab was initially approved for treatment of metastatic colorectal cancer in combination with chemotherapy, and its indications now include metastatic breast cancer, non-small-cell lung cancer, glioblastoma, renal cell carcinoma, ovarian cancer, and cervical cancer[7].
3 Cancer vaccines
Vaccination represents one of the most effective methods of preventing disease[2,8]. Preventive vaccines are designed to block transmission of disease and stimulate antibody production, B-cell memory and T-cell activation. Therapeutic vaccines are designed to eliminate cancer cells after they have formed. Common vaccine candidates use synthetic peptides, recombinant proteins, modified tumor cells, viral vectors, bacteria, and nucleic acids. These vaccines should be able to prime naïve T-cells and reprogram existing memory T-cells to give rise to tumor specific cytotoxic T lymphocytes(CTLs)[2]. Examples of therapeutic vaccines are:
• Cancer peptide vaccines—Cancer peptides are taken up by antigen presenting cells and stimulate an adaptive immune response. Free peptides are rapidly cleared before they can be loaded onto dendritic cells(DCs), thus will not trigger strong immune response. An example of an effort to stimulate a strong immune response involves co-administration of interleukin(IL)-2 with peptide derived from melanoma antigen glycoprotein 100(gp100)[9].
• Cell-based vaccines—GVAX is an example of a tumor cell-based vaccine, which uses modified tumor cells overexpressing granulocyte-macrophage colony-stimulating factor(GM-CSF). GM-CSF works in a paracrine manner to attract and active antigen-presenting cells[10].
• Nucleic acid-based vaccines—DNA,RNA, and viral vector-based vaccines deliver antigen or antigen fragments by an expression cassette. They transfect somatic cells or DCs, which lead to subsequent cross-priming or direct antigen presentation(respectively)[11]. DCs can be generated ex vivo, by isolating monocytes that are stimulated with cytokines such as GM-CSF, interleukins and interferons[12].
• DC vaccines—Mature DCs pulsed with antigens can be administered back into the patients[13]. Ex vivo generated DC vaccines enhance T-cell activation but their success has been limited by a variety of problems such as poor antigen presentation due to impaired lymph node trafficking[10].
4 CAR-T cells
T cells are thought to be the most important immune cells for cancer control. Chimeric antigen receptors(CARs) are fusion proteins that are introduced into T cells through gene transfer. The antigen- binding domain often consists of a mouse-derived monoclonal antibody as a continuous peptide single-chain variable fragment(scFv). The scFvs engage with their targets, such as tumor-associated antigens(TAAs), and activate T cell through the intracellular signaling domains[14]. CAR targeting allows binding to tumor antigen, and has no MHC restriction, and avoids many of the T cell escape mechanisms that are used by infectious agents and tumors[15]. CAR-T cells targeting the pan-B-cell marker CD19 have shown unprecedented response rates in treating refractory B cell lymphoma[16], and FDA has approved three products(tisagenlecleucel, axicabtagene ciloleucel and brexucabtagene autoleucel)[14].
Many tumors do not express unique tumor antigen, thus require the co-presence of two different antigens to activate the CAR-T cells. This approach increases specification and reduces the risk of either off-target recognition or “on-target, off-tumor” toxicities, in which healthy tissues that express the same antigen as tumor cells suffer collateral damage. However, the development of effective CAR-T cell therapy for non-B-cell malignancies has required more sophisticated engineering approaches to overcome tumor-defense mechanisms such as immunosuppression, antigen escape, and physical barriers to entry into solid tumors[17]. Cells other than T cells can be used with CARs.As macrophage have both cell killing and antigen presenting function, CAR-macrophage(CAR-M) has the potential to be more effective in stimulating adaptive anti-tumor response. CAR-natural killer(NK) cells can be generated from cord blood or iPSCs, and they have not MHC restricted, making them a potential allogeneic, off-the-shelf product[17].
5 Cytokines
Cytokines are small polypeptides or glycoproteins that regulate innate and adaptive immunity through mediating cell-to-cell communication[18]. Cytokines are most often released during a defined period in response to a stimulus, and the extent of their action is short-lived due to their limited half-life in the circulation. As a result, cytokines normally exert an autocrine or paracrine effect. Several cytokines limit tumor cell growth by a direct anti-proliferative or pro-apoptotic activity, or indirectly by stimulating the cytotoxic activity of immune cells against tumor cells. There are currently two cytokines that have been approved by FDA: IL-2 for metastatic melanoma and renal cell carcinoma, and IFN-α for Stage Ⅲ melanoma[19]. When used as monotherapy, cytokines exhibit low response rate and high toxicity, especially with high-dose IL-2. Current clinical trials are exploring combination therapies with a cytokine and checkpoint inhibitors(CPIs) or CAR-T cells. Fusion proteins using cytokines and antibodies, also known as “immunocytokines”, increase cytokine concentration in the tumor microenvironment(TME)[20]. An alternative strategy to achieve high local concentrations of cytokines in the TME is to directly inject gene therapy vectors that encode the cytokine into the TME[18,21].
6 Immune checkpoint inhibitors(CPIs)
Immune checkpoints are a normal part of the immune system that prevents immune response from destroying healthy cells. Tumor cells express ligands that bind to checkpoint proteins on the surface of T-cells, sending an “off” signal to avoid being killed[22]. Immune checkpoint inhibitors are antibodies binding to the checkpoint proteins, thus take off the “break” on T-cell cytotoxicity. Intensive research efforts have been focused on CPIs since the Nobel Prize was awarded to Dr. James P. Allison and Dr. Tasuku Honjo for their discovery of αCTLA-4 and αPD-1/L1 in cancer treatment, respectively. Ipilimumab(αCTLA-4) has led to considerable improvement in overall survival for patients with metastatic melanoma since it was approved by FDA in 2011[23]. Three αPD-1 antibodies have been approved by FDA: pembrolizumab(Keytruda), nivolumab(Opdivo), and cemiplimab(Libtayo). And three αPD-L1 antibodies have been approved by FDA: atezolizumab(Tecentriq), durvalumab(Imfinzi), and avelumab(Bavencio)[24]. In contrast to recombinant IL-2 or IFN- α as mentioned above, CPIs have a more favorable safety profile[18]. Currently, based on the search results on www.clinicaltrials.gov, there are 2 492 clinical trials testing αPD-1/PD-L1 CPIs, and 500 clinical trials testing αCTLA-4 worldwide.
7 Characteristics of cold tumors
ICPs have changed the treatment landscape of many tumors. However, in many cases the response rates have been modest. A major factor involving in initial resistance to ICP inhibitors is the lack or paucity of tumor T cell infiltration,which characterizes the so-called “cold tumors”[25]. In this part of the review, we describe potential mechanisms that lead to a “cold tumor”. Mechanisms of immune escape developed by inflamed tumors are reviewed elsewhere[26].
8 Lack of tumor antigens
Tumor elimination by T-cells starts with recognition of tumor antigens. Surface proteins expressed on tumor cells can mark them to be “nonself”. Tumor antigens can be divided into three main classes: tumor specific antigens(TSA), tumor associated antigens(TAA), and cancer-germline antigens(CGA)[27]. TSA are expressed only on cancer cells and not on normal cells[28]. TAA are expressed at low levels in normal tissue and overexpressed in cancer tissue. Both TSA and TAA are typically derived from intracellular molecules and get expressed on the cell surface as part of the major histocompatibility complex[29]. CGA are a class of immunogenic tumor antigens encoded by genes expressed in gametogenic cells of the testis and/or ovary and in human cancer[30]. Tumor antigens activate effectors, such as T cells, macrophages, and natural killer cells, which have tumoricidal abilities[29].
Tumors result from mutations. Tumor mutation burden(TMB) correlates with extent of immune infiltration[31]. In colorectal cancer, TMB predicts response to PD-1 treatment[27], but high TMB is not a good predictive biomarker for response to CPIs across all cancer types[32]. Furthermore, tumors with high TMB are not necessarily “hot”. In melanoma, there was no significant correlation between T-cell infiltration signature and presence of tumor antigens[33], and in the same study, Spranger et al examined T-cell signature across The Cancer Genome Atlas and found no correlation between T-cell gene expression and mutational burden in any cancer type.
9 Defects in antigen presentation
Major histocompatibility complex(MHC) proteins are necessary for the presentation of peptide antigens to cytotoxic T-lymphocytes(CTLs) and for the immune regulatory activity exerted by NK cells[34]. CD8+cells recognize abnormal/mutated proteins presented on MHC I to identify tumor cells and activate subsequent cytotoxicity. Since MHC I molecules are not essential for cell survival, one mechanism for tumor cells to evade immune surveillance is to down regulate expression of MHC I[35]. However, low MHC I expression leads to increased natural killer(NK) cell activation. But tumors evolve other mechanisms to combat NK cytotoxicity. For example, tumors often produce factors, such as transforming growth factor β(TGFβ) and prostaglandin that impair NK cell function and block their infiltration into the tumor site[36].
10 Disruption of normal chemokine
Although many tumors are eliminated by the immune system(and thus are never detected clinically), others continue to grow despite the presence of tumor antigens and generation of tumor-reactive T cells. Chemokines play a critical role in T cell trafficking and the specialization of immune responses[37]. Tumors can modify chemokine expression to suppress T-cells and keep them from reaching the tumor site. For example, human keratinocyte-derived skin tumors can regulate keratinocyte-specific chemokine CCL27 through the activation of epidermal growth factor receptor(EGFR)-Ras-MAPK-signaling pathways[38]. Basal and squamous cell carcinomas show limited mRNA and protein expression of CCL27 when compared to healthy skin. In an in vivo mouse tumor model, neutralizing CCL27 led to decreased CD4, CD8 and interferon-gamma(IFNγ) signature in tumor. Affymetrix gene expression profiling showed strong correlation between T cell associated markers and chemokine expression in metastatic melanoma[28]. Chemokine blockade with specific antibodies inhibited migration of CD8+T cells.
11 Vasculature barrier
In some tumors, we see T cells accumulate in the tumor stroma, but they fail to penetrate deeply into the tumor[37]. Tumor endothelial cells regulate immune cell migration, adhesion, and movement across the vessel wall. Antitumor immunity can be blocked by defects in the ability of leukocyte to adhere to tumor blood[39]. Angiogenic endothelial cells expressing high level of vascular endothelial growth factor(VEGF) inhibit lymphocyte adhesion through attenuating intercellular adhesion molecule-1(ICAM-1) and vascular cell adhesion molecule(VCAM-1) clustering. Overexpression of caveolae structural protein, caveolin-1, overcomes the VEGF-mediated inhibition of adhesion and restoreds ICAM-1 clustering, while knocking down caveolin-1 reduces the TNFα-dependent adhesion.
VEGF induces NO production, which results in cytoskeletal alterations that can lead to alterations in adhesion molecule clustering[39]. In human ovarian cancers, expression profiling shows that high endothelin B receptor[ET(B)R] is associated with the absence of TILs and short patient survival time[40]. Inhibition of ET(B)R increases T cell adhesion to human endothelium in vitro and can be reversed by ICAM-1 blockade or treatment with NO. These results suggest that ET(B)R mediates the endothelial barrier to T cell homing to tumors[40].
12 Attenuating the effectiveness of T-cell-mediated attack
T cell activation induces an inhibitory pathway that eventually attenuates and terminates T cell responses[23]. Recognition of antigen-MHC complexes by the T cell antigen receptor is not sufficient for activation of naïve T cells—additional costimulatory signals are required that are provided by the engagement of CD28 on the T cell surface with B7 molecules on APC. CTLA-4, which is on T cell surface, is first thought to be a costimulatory molecule for CD28. It is subsequently showed that CTLA-4 opposes CD28 costimulation and down-regulates T cell responses. Dr. James Allison was among the first scientists who discovered that CTLA-4 functioned as a brake on T cells. When CTLA-4 binds B7 molecules on APC, it leads to T cell accumulation at the T cell-APC interface, ultimately blocking costimulation and overturning an active T cell response[23](Figure 2).
PD-1 is mainly expressed on activated CD4+T cells and CD8+T cells, but is expressed on B cells in the periphery. The activation-induced expression of PD-1 suggests that PD-1 regulates late-phase immune responses important for memory response and controlling chronic infection. PD-1 was discovered in 1992 by Ishida et al. who isolated the gene that encoded PD-1 by cDNA subtraction in apoptosis-induced murine T-cell lines[41]. Like B7 molecules, PD-L1 is upregulated upon activation of APCs. PD-L1 is also expressed on activated T cells, vascular endothelial cells, various tumor cell lines and tumor tissues both in human and mouse[42]. Binding PD-1 to its ligands PD-L1/L2 leads to inactivation of T-cell receptor(TCR)-mediated signaling and inhibition of T cell proliferation and cytokine production such as IL-2 and IFNγ[41](Figure 2).
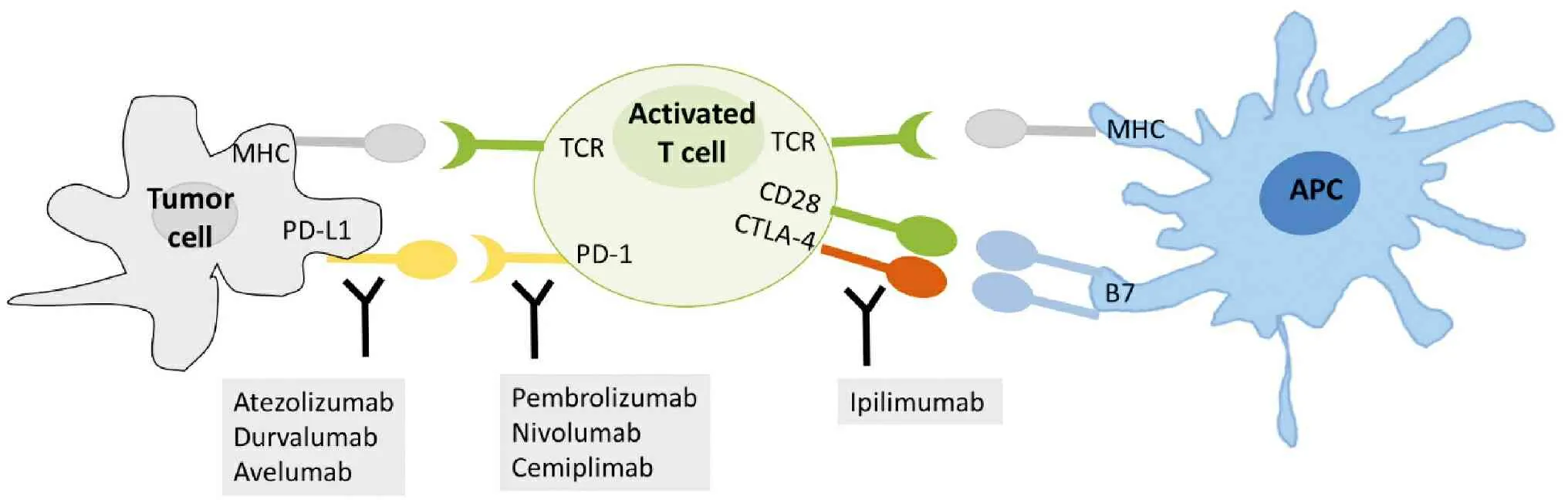
Figure 2 Illustration of cellular location of immune checkpoint proteins and FDA approved CPIs
13 Immune suppression regulators
Apart from immune checkpoint proteins, there are other suppressive factors of the immune response. T regulatory(Treg) cells and myeloid-derived suppressor cells(MDSCs) play important roles in inhibiting T effector cell function. Treg cells are CD4+CD25+FoxP3+and produce cytokines such as TGFβ, IL-10 and IL-35, which can suppress effector T cell expansion and suppress secretion of cytokines, such as IFNγ and TNFα[37]. Normally functioning Treg cells are important regulators that prevent autoimmunity. However, tumors can actively recruit Treg cells and Treg cells are associated with poor prognosis. Tumor-specific Tregs have been isolated from melanoma patients,which can be induced to expand after cancer vaccine treatment, and lead to attenuated T effector cell function[43]. Treg cells also express high CTLA-4 on their surface, which makes them a target for αCTLA-4[44]. Thus, CPIs promote T effector cell function directly, but they function indirectly by depletion of suppressing Treg cells.
14 Turning cold tumors hot
There are strong correlations between prolonged patient survival and the presence of intratumoral CD8+T-cells and IFNγ gene signatures[45]. The lymphocyte infiltration and IFNγ status may also be key factors for effective anti-PD-1/-L1 therapy and define a “T cell inflamed” phenotype(“hot tumors”). Here we discuss therapeutics with potential for turning cold tumors hot, improving the effectiveness of CPIs.
15 Specific therapy for tumor with low antigen or poor cross presentation
Epigenetic therapies regulate gene expression to increase tumor antigens[46]. Hypomethylating agents(HMA) such as azacitidine and decitabine are FDA approved drugs to treat acute myeloid leukemia. Novel HMA, like SGI-110,can not only increase TSA expression,but also increase MHC I expression, thus potentially contributing to the enhanced tumor recognition by cytotoxic T-cells[47].
MHC I expression can be restored through IFNγ treatment, leading to a partial restoration of sensitivity to CTL killing[48]. In vitro data showed that IFNγ treatment increased MHC I expression at 24 h regardless of baseline MHC I level. Although IFNγ treatment decreased NK cell killing of tumor cells, CTL-mediated lysis was increased significantly, thus leading to improved tumor-free survival in mice[48]. In a phase 0 clinical trial, weekly systemic IFNγ treatment significantly changed the patients′ TME, with increasing MHC I expression and T-cell infiltration. Some patients showed increased expression of PD-L1 on tumor-infiltrating myeloid cells and tumor cells, providing a rational for combining IFNγ and CPIs[49].
IL-12R activation results in activation of the JAK-STAT pathway, which ultimately leads to IFNγ production, transcriptional reprogramming of CD4+lymphocytes towards type 1 T helper(Th1) cell differentiation and maturation of NK cells[18]. IFNα has anti-tumor activities and can directly kill tumor cells and stimulate DCs and CD8+T cells[50]. A fusion protein of IL-12 with antibody against tumor antigen led to increased tumor infiltrating lymphocytes, macrophages, and natural killer cells, which in turn increased IFNγ[51]. Induction of IL-12 and IFNα turns the immune suppressive tumor microenvironment into an immune supportive environment. Synthetic peptides vaccines can be conjugated to TLR ligands such as CpG to promote responsive DCs to secrete IL-12 and IFNα[52]. Mouse studies showed that tumor antigens complexed with β-glucan injected in combination with CpG, induced potent CTL activity and demonstrated delayed tumor growth[53]. The author pointed out that peptide alone was not a good vaccine. In a mouse melanoma model, DCs pulsed with a low dose of liposomal tumor peptide and CpG improved PD-1 blockade immunotherapy when compared with DC pulsed with peptide alone[54]. The CD8+T effector to Treg cell ratio, cytotoxic lysis and IFNγ were the highest in the liposomal peptide plus CpG group.
16 Increasing homing and infiltration of TILs
An early study showed that αPD-1 alone enhanced recruitment of effector T cells and inhibited the hematogenous spread of poorly immunogenic B16 melanoma and CT26 colon cancer cells[42]. While there was no PD-L1 expression in vitro, B16 cells started to express PD-L1 after inoculation into mouse liver to establish tumors. Compared with that in wild-type mice, intrasplenic injection of B16 cells in PD-1 deficient mice showed increased effector T cells in spleen, prolonged T cell proliferation and cytokine production. These results show that PD-1 blockade increases homing of effector T cells to tumor[42].
PD-1 blockade monotherapy does not always deliver satisfactory outcome. Finding reliable makers to predict response to CPIs is important for patients who may not benefit from the treatment or suffer from side effects. In pre-clinical models, VCAM-1 density could be used to predict T-cell infiltration and PD-L1 treatment response[55]. Microparticles of iron oxide(MPIO) have been conjugated to VCAM-1 antibody for MRI detection of tumor VCAM-1 density.In this mouse colon cancer model,VCAM-MPIO tumor binding and perfusion correlated with T cell infiltration. As expected, antibodies blocking VCAM-1 and ICAM-1 decrease T cell binding to endothelial cell, thus preventing tumor rejection. Baseline VCAM-MPIO tumor binding and perfusion predicted αPD-L1 therapy outcome[55].
Because VCAM-1 controls leukocyte adhesion and migration along endothelial cells(ECs), it may be more than a predictive marker for CPIs. In a human glioblastoma(GBM) model, p21-activated kinase 4(PAK4) was shown to be a critical regulator of GBM ECs[56]. EC-specific PAK4 knockout showed reduced EC abnormalities, improved T cell infiltration, and inhibited GBM. Moreover, pharmacologic inhibition of PAK4 showed reduced proliferation of GBM ECs and normalized the tumor vasculature.Low PAK4 led to increased VCAM-1 and ICAM-1 expression and enhanced T cell binding to the GBM ECs. PAK4 inhibition resulted in improved CAR-T treatment for GBM[56].
17 Improving cytotoxic function of TILs
Prostate cancer is immunologically “cold”, and rarely responses to CPIs. Based on the observation that hypoxia down-regulated T cell adhesion molecules and induced T cell apoptosis, a combination therapy of hypoxia-activated prodrug TH-302 and CPIs was evaluated in a mouse prostate cancer model[57].TH-302 treatment significantly reduced hypoxic areas in prostate cancer, increased survival, and suppressed tumor growth. In mice receiving TH-302 with CPIs, CD8+T cell proliferation(Ki67), cytotoxic potential(granzyme B), activation(CD44), and effector cytokine production(IFNγ and TNFα) all increased. At the same time, proliferation of granulocytic MDSCs in hypoxic zones of prostate tumors decreased, as did their capacity to suppress T cell proliferation[57].
Another approach to combat prostate cancer resistance to CPIs involves modifying the regulator for interferon-stimulated genes(ISGs). EZH2 negatively regulates ISGs, including expression of Th1-type chemokines, PD-L1 and MHC in multiple cell types. In a mouse prostate cancer tumor model, combination of EZH2 inhibition and αPD-1 significantly reduced tumor growth, with upregulation of PD-L1 that was associated with improved αPD-1 response and increased immune-mediated cytotoxicity[58]. EZH2 inhibition also resulted in increased infiltration of CD3+, CD4+and CD8+cells into tumor with upregulation of Th1 chemokines IL-2, IL-12 and TNFα. In addition, combination of EZH2 inhibition and αPD-1 reversed the immune suppressive TME by increasing intratumoral M1 tumor-associated macrophages(TAMs) and decreasing M2 TAMs, thus increasing the M1:M2 TAM ratio in prostate cancer[58]. Similarly, in a melanoma model, depletion of EZH2 inhibition led to robust antitumor immunity, with decrease in Treg cells and enhancement of cytotoxic activity of T effectors[59]. In combination with αCTLA-4 therapy, EZH2 inhibition further reduced tumor growth, increased survival when compare to either of the monotherapy.
PAK4 inhibition is another candidate to improve CPIs in melanoma.Expression profiling showed that CD8+cell infiltration in melanoma patients was anti-correlated with PAK4 levels, with the αPD-1 responders showing increased CD8 cytotoxicity[60]. In a mouse model, PAK4 knockout led to improved αPD-1 response and increased immune cell infiltration. These results suggest that inhibition of PAK4 primes the tumor to be more sensitive to αPD-1 and CD8-dependent anti-tumor immunity. An analysis of transcriptome data from The Cancer Genome Atlas showed that PAK4 was negatively correlated with immune cell infiltration across human cancer types, thus making PAK4 inhibition a promising candidate for improving CPI response.
18 Conclusion
There is an urgent effort to develop clinical trials for therapies that propose to increase immune infiltration in cold tumors. Some of the pre-clinical models discussed above have led to clinical trials. Examples include NCT03525795(EZH2 inhibition with αCTLA-4) and NCT02702492(PAK4 inhibition with αPD-1). These studies are expected to shed light on the efficacy and safety of immunotherapies for cold tumors. In the near future, immunotherapies may become the mainstay of treatment for all tumors.
