
极小种群野生植物海南风吹楠的遗传多样性研究
2021-10-13 11:18:38蔡超男侯勤曦慈秀芹肖建华张灿瑜李捷
热带亚热带植物学报 2021年5期
蔡超男, 侯勤曦, 慈秀芹, 肖建华, 张灿瑜, 李捷*
极小种群野生植物海南风吹楠的遗传多样性研究
蔡超男1,2, 侯勤曦3, 慈秀芹1,4, 肖建华1,5, 张灿瑜1,5, 李捷1,4*
(1. 中国科学院西双版纳热带植物园,昆明 650223; 2. 台州学院高等研究院, 浙江 台州 318000;3. 四川省大熊猫科学研究院,成都 610081; 4. 中国科学院核心植物园,云南 勐腊 666303; 5. 中国科学院大学,北京 100049)
为探讨海南风吹楠()的濒危原因,利用限制性酶切位点相关的DNA测序技术(RAD-seq)开发单核苷酸多态性(SNPs),评估居群的遗传多样性和遗传结构。结果表明, 海南风吹楠的遗传多样性较低(=0.167),其中BWL居群表现出最高的遗传多样性;居群间存在中等程度的遗传分化(F=0.120)。Structure分析表明居群的最佳聚类值为2,但个别居群的遗传结构混杂,Mantel检测结果也表明遗传距离和地理距离没有相关性(=0.733,<0.075)。自身更新能力低以及过度的人为活动干扰,可能是导致其濒危的主要原因。建议加强对遗传多样性高的居群(BWL和YGL)进行就地保护; 对生境破坏严重的居群(EXL和DLS)进行近地或迁地保护,以增加居群间的基因交流,同时构建核心种质,防止遗传资源丢失加剧。
海南风吹楠;RAD-seq;遗传多样性;极小种群;遗传结构
随着人类社会和科学技术的进步与发展,全球气候持续发生变化,很多物种处于濒危状态,生物多样性正以前所未有的速度丧失,甚至正在推动地球历史上的第六次大规模灭绝,因此对生物多样性保护制定合理高效的保护计划显得尤为重要和迫切[1–3]。当前物种保护最突出和最具争议的一个问题是:由于保护所有的濒危物种非常困难,哪些物种才是最需要优先保护的?2005年云南省林业厅首次在国内提出“极小种群野生植物(plant species with extremely small population, PSESP)”的概念,是指在特殊地区的特定环境下长期形成的、分布区域狭窄或呈不连续分布,由于物种本身的因素或长期受到外界胁迫因素的干扰,种群和个体的数量不断减少已经低于物种稳定存活的最小生存种群,难以维系物种的正常繁衍而随时濒临灭绝的植物种类[4–5]。经过十几年的不断发展和完善,极小种群野生植物的概念已得到国家和科学界的认可。
海南风吹楠()隶属于肉豆蔻科(Myristicaceae)风吹楠属,是热带雨林的标志物种,且具有很高的经济和药用价值,主要分布在海南省[6–8]。由于岛上天然林被大量砍伐,加上居民的保护意识薄弱等原因,当前被列入全国极小种群野生植物物种名录中[9],说明对该物种的保护工作已迫在眉睫。物种保护的核心是要尽可能地保护其遗传变异水平,它是物种适应复杂多变环境的基础,一个物种的种内遗传多样性越高或遗传变异越丰富,物种的进化潜力及对环境变化的适应能力就越强,因此对海南风吹楠开展保护遗传学研究对于制定有效的保护策略至关重要[10–11]。
近年来,有关海南风吹楠的基础研究主要集中在种子萌发、生理特性、生物学特性以及群落区系特征等方面[12–14],居群的遗传多样性研究尚处于起步阶段[15]。物种的正确界定是了解、保护和利用生物资源的前提[16–17]。Cai等[18]的研究表明,海南风吹楠仅分布于海南省,而Jiang等[15]基于简单序列重复区间(inter-simple sequence repeat, ISSR)分子标记对海南风吹楠开展的遗传多样性研究,将分布在广西地区的滇南风吹楠认定为海南风吹楠,因此不能正确评估海南风吹楠的遗传多样性水平。此外, 采用的ISSR分子标记不能区分显性纯合和显性杂合基因以及筛选出的核苷酸位点仅能覆盖基因组的一小部分,导致对海南风吹楠的遗传多样性评估不准确[19–20]。与ISSR、简单重复序列(simple sequence repeats, SSR)等传统分子标记以DNA片段的长度变化作为检测手段不同,单核苷酸多态性(single nucleotide polymorphisms, SNP)直接以序列变异作为标记[21]。SNP标记的主要优点是其在全基因组分布均匀且数目多能够稳定遗传,检测容易、易实现自动化[22]。新一代DNA测序技术的应用,特别是限制性酶切位点相关的DNA测序技术(restriction- site associated DNA sequencing, RAD-seq)的出现, 使得低成本、耗时短及高通量开发非模式物种的SNP分子标记更为便捷,使得越来越多的研究者利用该技术对物种进行保护遗传学研究[23–25]。
因此,为对极小种群野生植物的保护提出合理可靠的建议,在准确把握物种界定后,利用新的测序技术手段开发SNPs标记对海南风吹楠进行保护遗传学等相关研究是十分必要的。本研究利用RAD-seq技术对极小种群野生植物海南风吹楠进行遗传多样性评估和遗传结构的探讨,以阐明该物种的种质遗传背景,进而提出具有针对性的保护策略。
1 材料和方法
1.1 样品采集
于2017年5月在海南省采集海南风吹楠5个自然居群的新鲜、幼嫩叶片,用硅胶迅速干燥。每份材料距离间隔尽量大于100 m,共50份材料(表1,图1),标本存放于中国科学院西双版纳热带植物园标本馆(HITBC)。
1.2 总DNA提取
野外采集经硅胶干燥后的新鲜叶片存储于–20℃冰箱。利用4×CTAB法提取植物总DNA[26]。检测合格的DNA样品(总量≥1g,浓度≥20/L)送到深圳华大基因科技服务有限公司(BGI)进行测序。因该物种的DNA提取存在一定困难,最终根据测序质量要求仅获得5个居群共20株个体(图1, 表2)。
1.3 DNA文库构建和测序
RAD-seq参考Davey等[27]的方法。用R I限制性内切酶(5ʹ-GAATTC-3ʹ)处理基因组DNA,得到具有粘性末端的酶切片段。在酶切后DNA两端加上P1接头并机械打断,电泳回收目的片段;在酶切片段的另一端加上P2接头,用P1、P2引物进行PCR扩增,回收目的片段;对检测合格的样品进行上机测序,采用Illumina Hiseq 2000进行双端测序(PE=150 bp)。样品的文库构建和测序都在深圳华大基因科技服务有限公司完成。
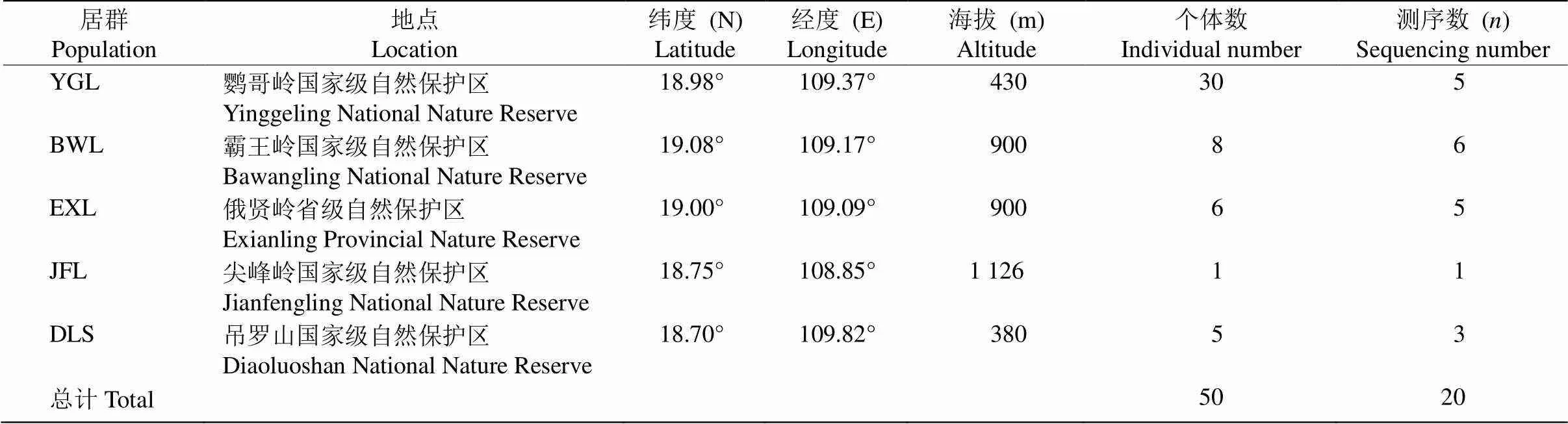
表1 海南风吹楠采样信息
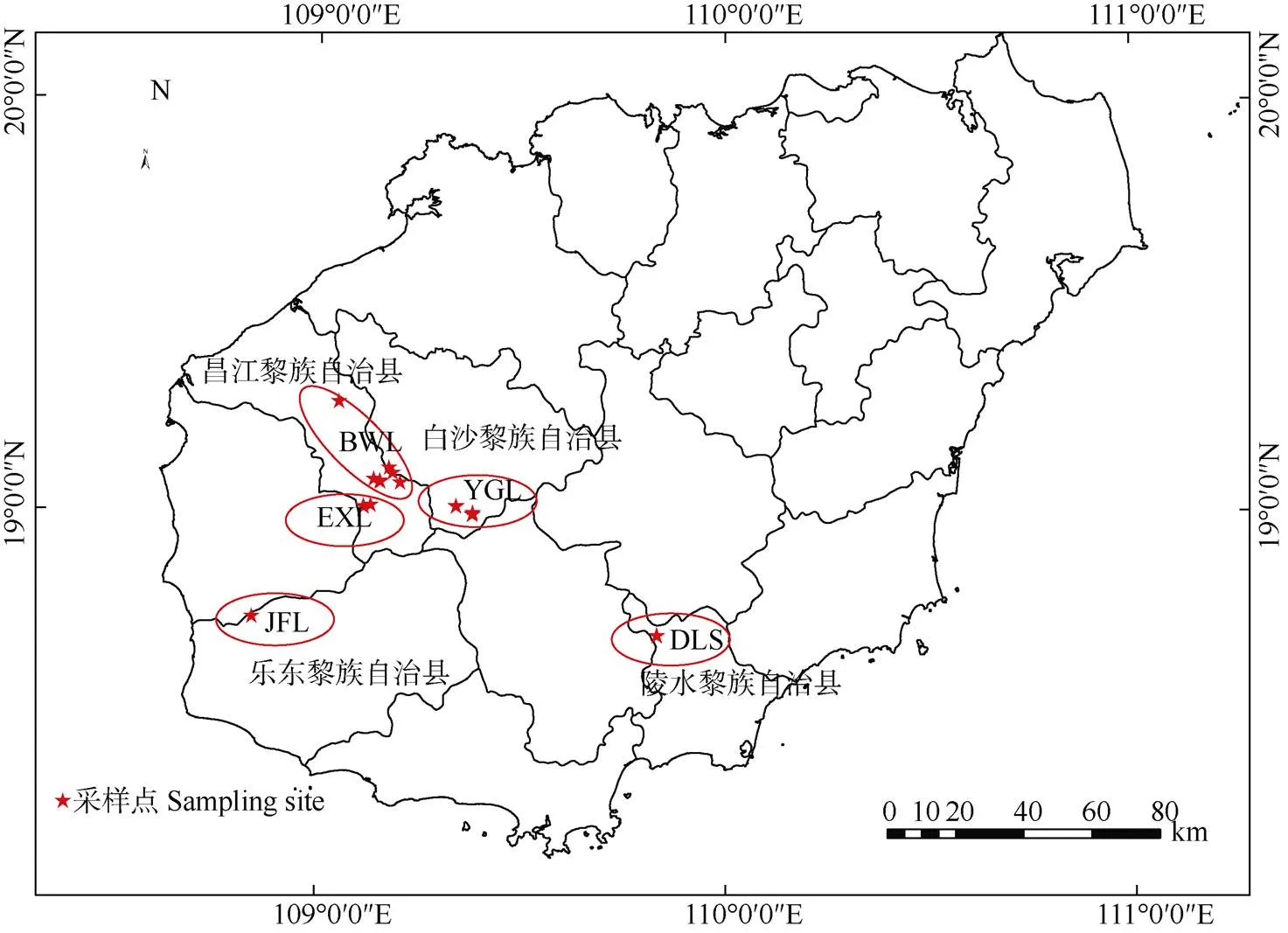
图1 海南风吹楠的采样点
1.4 个体聚类与居群聚类
首先对原始数据进行质量过滤,统计reads数、Q值(单碱基错误率)、GC含量等参数,保留Q20值达到98%以上的数据。利用软件Stacks v.2.2[28]对数据进行分析处理。首先,使用对所有样本的1’reads进行聚类,将深度在3以上的相似序列堆列在一起,形成1个stack,允许生成stack的两条序列至多有2个碱基的差异(即设置参数m=3和M=2);随后利用通过比对,生成个体间的一致性位点(consensus loci),并将信息写入目录文件catalog (设置参数n=2);然后利用将由生成的stack再重新匹配到catalog文件,以确定个体内相应位点的基因型。接着执行程序,以便将数据从按样本聚类转换到按RAD位点进行聚类,得到位点比对矩阵(i.e: *.bam)。利用整合双端序列(1’reads和2’reads)形成contig,然后将contig比对到locus上,并在群体水平call变异以及分型,找出每个个体SNPs。
1.5 遗传多样性分析
利用软件Stacks v.2.2[28]中的程序进行SNPs过滤及获取遗传多样性参数。为挖掘SNPs分子标记,设置参数:--write_single_snps、r=0.8、p=3。--write_single_snps表明仅输出第一个SNPs位点作为后续遗传多样性分析的标记;r为0.8表明该位点在某一居群中至少有80%的个体共享; p为3表明该位点至少被3个居群共享。为了评估物种水平上的遗传多样性,将20个海南风吹楠样品作为1个居群来处理,并要求位点在居群中至少为80%的个体共享(p=1, r=0.8)。利用SNPs标记评估居群的遗传多样性,计算以下参数:在所有位点和变异位点中的私有等位基因数(private allele number,)、期望杂合度(expected heterozygosity,)、观测杂合度(observed heterozygosity,)及近交系数(inbreeding coefficient,F)等。
利用PGDSpider v 2.1.1.5[29]软件对文件格式进行转换。采用Arelequin v 3.5[30]软件进行分子方差分析(analysis of molecular variance, AMOVA),计算遗传变异在居群间和居群内的分布及居群间的遗传分化系数(F)以及物种水平上的基因流()= (1–F)/4F[31]。使用软件Stacks v.2.2[28]中的程序计算居群间的F值,居群间的地理距离通过居群的地理坐标信息进行计算。在R环境中使用vegan包的“Mantel”函数[32]检验居群的遗传距离与地理距离的相关性。
1.6 群体的遗传结构分析
利用软件Structure v. 2.3.4[33]进行群体的遗传结构分析。数据分析时选择混合模型(admixture model)和等位基因频率相关模型(allele frequencies corre- lated),程序的参数“length of burn-in-period”设定为1 000 000, “number of MCMC replications after burnin”设定为2 000 000,K值设定为1~5,迭代次数设定为10次。运行结果压缩后上传至软件Structure Har- vester v. 0.6.94[34]进行分析,然后利用软件CLUMPP v. 1.1.2[35]和DISTRUCT v. 1.1[36]将最佳K值的10次重复运行的结果整合起来,生成群体遗传结构图。
2 结果和分析
2.1 测序数据与RAD位点数
从表2可见,海南风吹楠序列的原始reads数(clean reads)为6 894 604~40 535 670,个体平均为17 855 431。测序平均Q20为98.6%,平均GC含量为40.8%。为确保某1位点至少被3个居群的80%个体共享,评估居群遗传多样性时共保留142 140个RAD位点,得到11 225个SNPs用于后续的遗传多样性和群体结构分析。
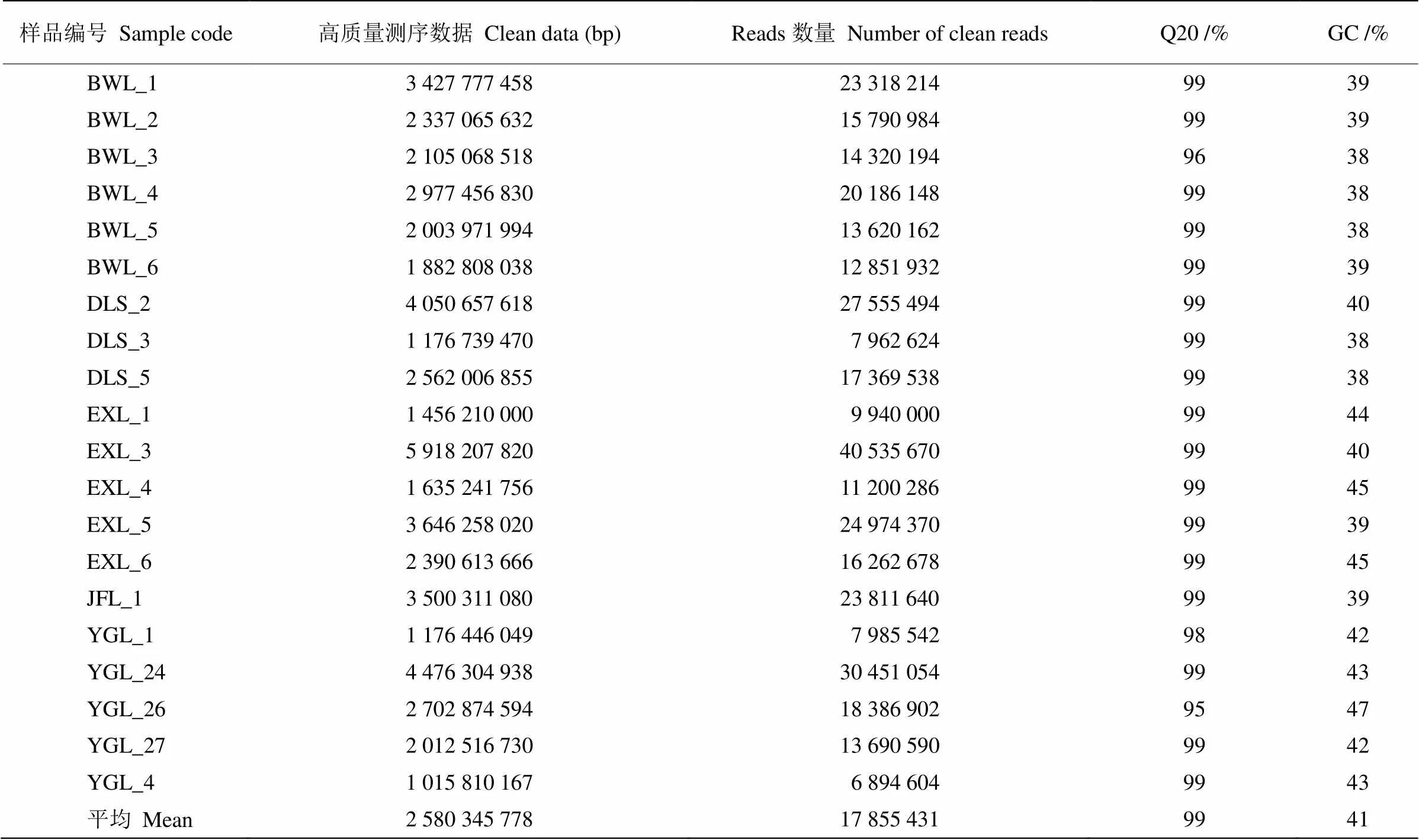
表2 海南风吹楠的RAD测序数据
2.2 遗传多样性
从表3可见,物种水平上,极小种群野生植物海南风吹楠的观测杂合度()为0.151,期望杂合度()为0.167,核苷酸多样性()为0.172,近交系数(F)为0.081。居群水平上,海南风吹楠的为0.147~0.184,为0.075~0.175,为0.151~0.192,F为–0.014~0.045。同时,BWL居群表现出最高的遗传多样性。
2.3 居群遗传分化与遗传结构
从表4可见,海南风吹楠物种水平的F为0.081,群体间的F为0.070~0.230。尖峰岭(JFL)与吊罗山(DLS)居群的遗传分化系数最大,Mantel分析结果表明地理距离和遗传距离具有正相关性,但不显著(=0.733,<0.075, 图2)。AMOVA分析结果表明遗传变异主要分布在居群间(大约75.47%),只有24.53%的遗传变异源自居群内(表5)。居群遗传结构分析结果表明,当K=2时,ΔK散点曲线出现最大值,表明海南风吹楠群体的最佳聚类值为2,然而根据其地理分布,居群遗传结构分析无法明显地划分为2个组(图3),这可能是居群及个体数太少或者居群间距离太近导致的。
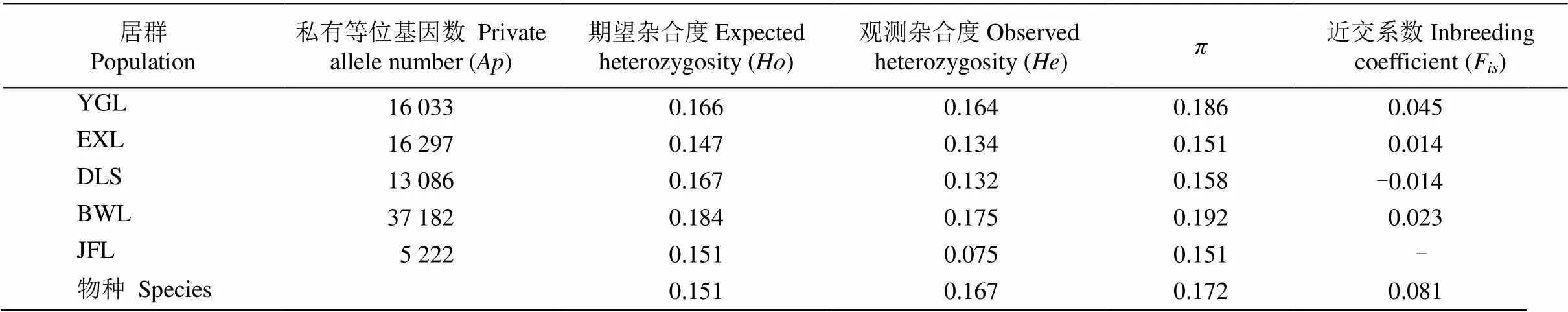
表3 海南风吹楠物种和居群水平的遗传多样性
YGL、EXL、DLS、BWL、JFL见表1。以下图表同。
YGL, EXL, DLS, BWL, JFL see Table 1. The same is followed Tables and Figures.
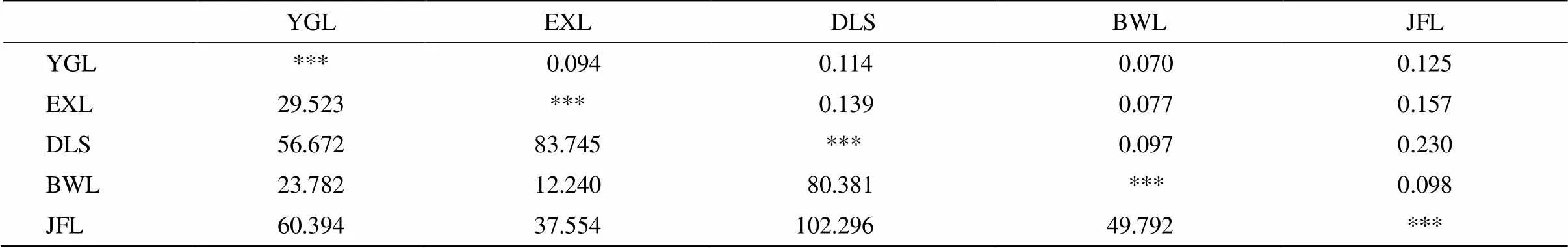
表4 海南风吹楠居群的遗传距离(Fst, 右上)与地理距离(km, 左下)
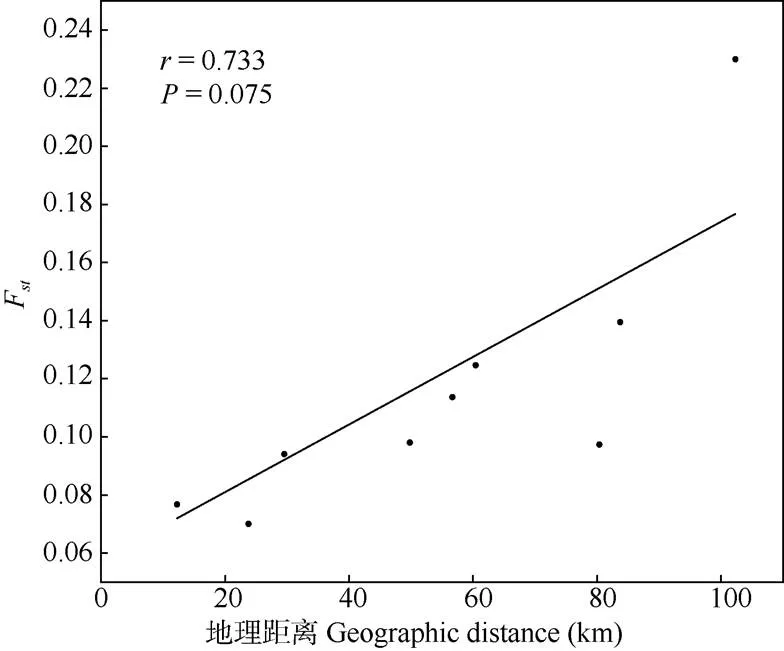
图2 海南风吹楠居群间的遗传距离(Fst)和地理距离(km)的Mantel检验
3 结论和讨论
3.1 海南风吹楠的遗传多样性
本研究利用RAD-seq技术分析了极小种群野生植物海南风吹楠5个居群共20个个体的遗传多样性,结果表明,遗传多样性()在物种水平(0.167) 和居群水平(0.136)上都较低。与濒危植物云南蓝果树(, 0.321)[38]相比,海南风吹楠有更低的遗传多样性。目前利用其他分子标记的研究也表明濒危和极小种群植物的遗传多样性低,用RAPD标记版纳青梅()的 0.169[39]、ISSR标记伯乐树() 的0.141[40],用AFLP标记琴叶风吹楠()的0.152[41]等。

表5 海南风吹楠居群间和居群内遗传变异的AMOVA分析
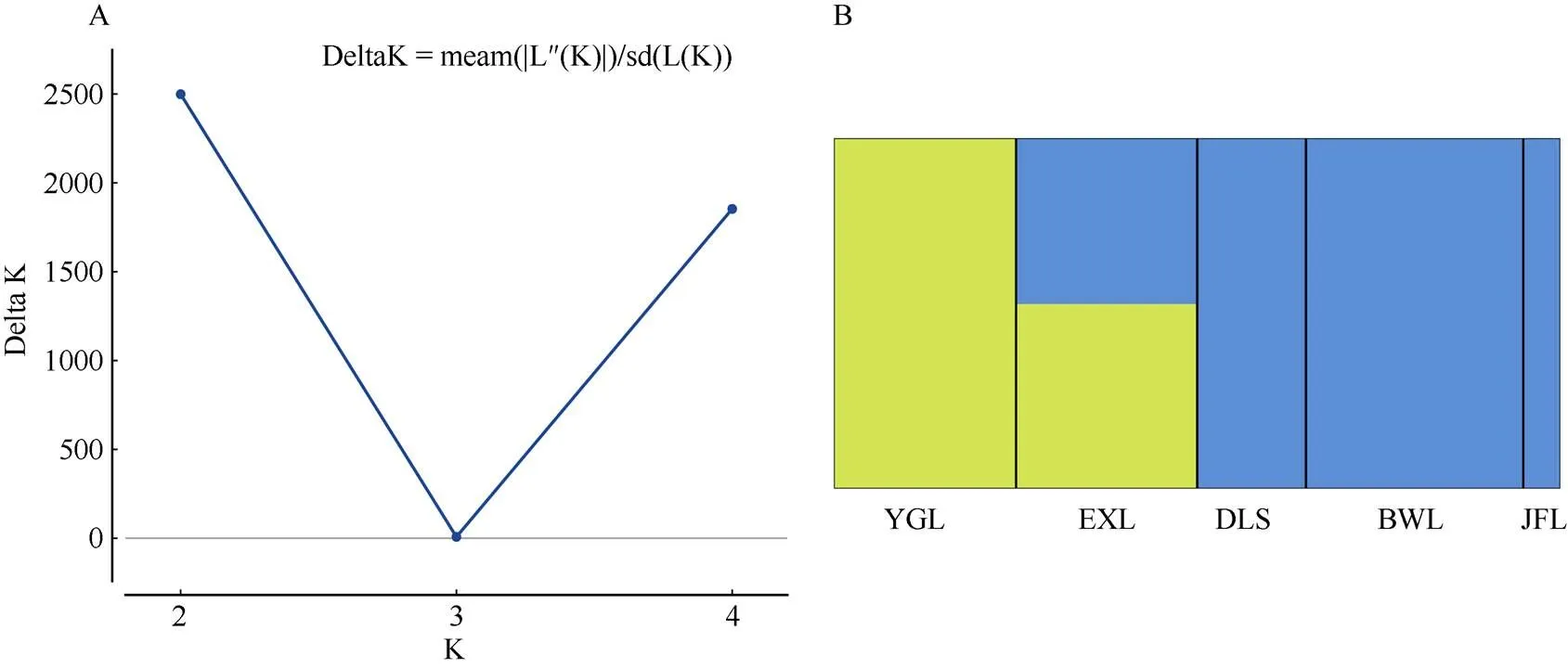
图3 基于Structure分析的海南风吹楠聚类的后验概率图(K=2)。A: 每个K值对应的delta K; B: K=2。
植物的遗传多样性受到多种因素的影响,是其进化历史、地理分布范围、繁殖方式等多种因素综合作用的结果[42]。本研究表明,海南风吹楠居群间的基因流有限(=0.081),且存在近交现象(F> 0),这可能是因为该物种主要生长在河谷或狭谷石缝的阴湿环境中,居群个体数少、林下幼苗及幼树少,在新的环境很难拓殖并扩大居群的规模,导致居群内近交衰退现象,物种表现出较低的遗传变异[42]。海南风吹楠的种子呈椭圆形,长约4.5 cm,直径约2.5~ 3 cm,主要靠重力传播,结合其种子较大导致其传播距离短,种子雨覆盖在母树周围,限制了群体的扩张。此外,热带雨林潮湿的环境容易致使种子腐烂,加上种子富含丰富的油脂容易引起虫蚁对种子的啃食等原因限制了它的萌发,导致在天然居群内其林下幼苗数量较少[12]。这些在很大程度上决定了海南风吹楠长期以来可能都是以小种群的形式存在,种群更新困难甚至停滞,从而限制了居群的扩张,导致遗传多样性下降。
另外,环境因素(生物因素、非生物因素和微环境等)的变化也会影响物种的遗传多样性水平[43]。日本占领海南岛期间,大肆掠夺木材,破坏原始森林;解放后,当地居民为了生活,砍伐木材,种植橡胶,全岛的森林覆盖率从解放初的35%,下降到20世纪80年代初的9.7%[44]。海南风吹楠可能本身就是个小种群植物,再加人为滥砍滥伐,导致海南风吹楠居群的规模变得更小,大多数地区只包含几棵植株, 促使居群内近交现象越发明显,可能因此导致物种的遗传多样性处于较低水平。综上,在本研究中, 海南风吹楠的遗传多样性低可能与其自身更新能力低以及人类对其生境的严重破坏以及滥砍滥伐有关,最终可能导致物种的局部消失与灭绝。
3.2 海南风吹楠的遗传分化与居群遗传结构
海南风吹楠居群间的平均遗传分化系数为0.120, 说明居群间存在中等程度的遗传分化[45]。AMOVA分析揭示了遗传变异主要存在于居群间, 遗传分化通常是长期遗传隔离的结果,受交配系统、生活史特征、传粉生物学、种子传播、生活型、气候的反应和基因流等生物学特性的影响[46]。在热带,对于混交的、虫媒传粉的非木本物种,F通常较高。海南风吹楠分布于热带地区,其雄花呈亮黄色,这是植物在吸引蜂类传粉者功能中呈现的主要色彩[47],所以我们推测海南风吹楠是靠蜂类传粉的。而蜂类传粉可能会导致海南风吹楠花粉流传播距离不长,居群间基因流交流困难,这可能导致居群间容易形成长期的隔离和分化[46]。
在野外科考时发现多数海南风吹楠居群间有山体隔离,未发现传播种子的鸟类,种子主要靠重力传播,传播距离有限,幼苗大部分聚集在母株下方区域。此外,人类频繁的活动使物种的生境遭到严重破坏(如霸王岭和吊罗山国家级自然保护区内海南风吹楠的一些分布点位于公路和栈道的两边),居群数量迅速减少,从而加剧了物种居群间的遗传分化[15,46,48]。
居群遗传结构分析结果表明,海南风吹楠居群的最佳聚类值是K=2 (图3),个别居群的遗传结构混杂,地理区域性特征不明显,Mantel检验结果表明遗传距离和地理距离虽呈正相关但不显著,两者的结果是一致的。Wang等[49]对分布在中国海南岛的濒危植物坡垒()的研究有相同的结果,坡垒居群的遗传结构混杂,地理距离和遗传距离间没有相关性,推断花粉的长距离基因流动、种子迁移以及地理距离等可能是导致该物种遗传结构不清晰的原因。此外,评价居群的遗传结构需要结合物种的繁育系统和传粉方式等因素进行综合分析[41],目前还没有针对海南风吹楠相关的报道,后续应加强对这方面的研究来进一步阐明其遗传结构的形成原因。
3.3 海南风吹楠的濒危机制及保护措施
居群结实率低、土壤种子库中种子储量小、幼苗死亡率高和过度的人为活动干扰等原因可能是海南风吹楠濒危的主要原因[12,50]。因此,我们提出对国家级极小种群野生植物海南风吹楠的科学保护措施,第一,掌握海南风吹楠的生存现状,针对性地开展繁育系统、传粉方式等研究,这些基础信息能为采取有效保护措施提供重要的科学依据;第二,加强对海南风吹楠天然居群的就地与迁地保护相结合的保护措施。散生于保护区外的海南风吹楠(如昌江王下乡农田边有1棵),其生境破坏严重, 急需进行迁地保护;位于保护区内的居群,对生境破坏的居群(如BWL和DLS居群)加强就地或近地保护,使其在原生或相似的生境区域逐渐扩大个体数量和居群规模;第三,加强对海南风吹楠的人工繁殖和回归引种措施。从遗传多样性水平高的居群(如BWL和YGL居群)中采集种子或幼苗,进行人工育苗后,选择适宜其生长的环境,以此扩大和更新现有居群的数量及规模,保护物种的基因库,防止物种的遗传资源发生丢失。此外,应该在当地加强宣传教育,最大限度的避免人为破坏导致野外居群数量的减少和生存环境的破坏。
[1] ISBEL F, GONZALEZ A, LOREAU M, et al. Linking the influence and dependence of people on biodiversity across scales [J]. Nature, 2017, 546(7656): 65–72. doi: 10.1038/nature22899.
[2] JOHNSON C N, BALMFORD A, BROOK B W, et al. Biodiversity losses and conservation responses in the Anthropocene [J]. Science, 2017, 356(6335): 270–275. doi: 10.1126/science.aam9317.
[3] LAWTON J H, MAY R M. Extinction Rates [M]. Oxford: Oxford University Press, 1995.
[4] REN H, ZHANG Q M, LU H F, et al. Wild plant species with extremely small populations require conservation and reintroduction in China [J]. AMBIO, 2012, 41(8): 913–917. doi: 10.1007/s13280-012-0284-3.
[5] MA Y P, CHEN G, GRUMBINE R E, et al. Conserving plant species with extremely small populations (PSESP) in China [J]. Biodiv Conserv, 2013, 22(3): 803–809. doi: 10.1007/s10531-013-0434-3.
[6] MERRILL E D. A fourth supplementary list of Hainan plants [J]. Lingnan Sci J, 1932, 111: 37–61.
[7] Institute of Medicinal Plant Development Hainan Branch. Species List of South China Medicinal Plant Garden [M]. Beijing: China Agriculture Press, 2007: 26.
中国医学科学院药用植物研究所海南分所. 南药园植物名录[M]. 北京: 中国农业出版社, 2007: 26.
[8] LIU S B, MEI W L, ZENG Y B, et al. Study on liposoluble extract of[J]. Chin J Ethnomed Ethnopharm, 2010, 19 (20): 147,149. doi: 10.3969/j.issn.1007-8517.2010.20.145.
刘寿柏, 梅文莉, 曾艳波, 等. 黎药海南风吹楠脂溶性成分研究[J]. 中国民族民间医药, 2010, 19(20): 147,149. doi: 10.3969/j.issn.1007- 8517.2010.20.145.
[9] State Forestry Administration of China. The Implementation Plan of Rescuing and Conserving China’s PSESP (2011–2015) [M]. Beijing: State Forestry Administration of China, 2012.
国家林业局. 全国极小种群野生植物拯救保护工程规划(2011种群野生植物) [M]. 北京: 国家林业局, 2012.
[10] GE S, HONG D Y. Genetic diversity and its detection method [M]// QIAN Y Q, MA K P. Principles and Methods of Biodiversity Research. Beijing: China Science and Technology Press, 1994: 122–140.
葛颂, 洪德元. 遗传多样性及其检测方法[M]// 钱迎倩, 马克平. 生物多样性研究的原理与方法. 北京: 中国科学技术出版社, 1994: 122–140.
[11] AVISE J C, HAMRICK J L. Conservation Genetics: Case Histories from Nature [M]. New York: Chapman & Hall, 1996.
[12] JIANG Y H. Ecological characteristics and endangered reason analysis ofMerr. as an extremely small population [D]. Changsha: Central South University of Forestry and Technology, 2018.
蒋迎红. 极小种群海南风吹楠生态学特性及濒危成因分析[D]. 长沙: 中南林业科技大学, 2018.
[13] YANG Y, LIU Q, CHEN Y K, et al. The complete chloroplast genome of, an endangered species with extremely small populations [J]. Mitochondrial DNA B, 2019, 4(2): 2654–2655. doi: 10.1080/23802359.2019.1644556.
[14] HUANG J, WANG H Y, ZHONG Y D, et al. Growth and physiological response of an endangered tree,Merr., to simulated sulfuric and nitric acid rain in southern China [J]. Plant Physiol Biochem, 2019, 144: 118–126. doi: 10.1016/j.plaphy.2019.09.029.
[15] JIANG Y H, LIU X S, XIANG W H, et al. Genetic diversity and structure analysis of the endangered plant speciesMerr. in China [J]. Biotechnol Biotechnol Equip, 2018, 32(1): 95–101. doi: 10.1080/13102818.2017.1391122
[16] DESALLE R, AMATO G. The expansion of conservation genetics [J]. Nat Rev Genet, 2004, 5(9): 702–712. doi: 10.1038/nrg1425.
[17] HONG D Y. Biodiversity pursuits need a scientific and operative species concept [J]. Biodiv Sci, 2016, 24(9): 979–999. doi: 10.17520/ biods.2016203.
洪德元. 生物多样性事业需要科学、可操作的物种概念 [J]. 生物多样性, 2016, 24(9): 979–999. doi: 10.17520/biods.2016203.
[18] CAI C N, MA H, CI X Q, et al. Comparative phylogenetic analyses of Chinese(Myristicaceae) using complete chloroplast genome sequences [J/OL]. J Syst Evol, (2019-12-12) doi: 10.1111/jse. 12556.
[19] KADU C A C, MUCHUGI A, KINDT R, et al. Molecular markers for tropical trees: A practical guide to principles and procedures [M]// DAWSON I, JAMNADAS R. ICRAF Technical Manual Vol. 9. Nai- robi: World Agroforestry Centre, 2008.
[20] ANGELONI F, WAGEMAKER N, VERGEER P, et al. Genomic toolboxes for conservation biologists [J]. Evol Appl, 2012, 5(2): 130– 143. doi: 10.1111/j.1752-4571.2011.00217.x.
[21] SEMAGN K, BJØRNSTAD Å, NDJIONDJOP M N. An overview of molecular marker methods for plants [J]. Afr J Biotechnol, 2006, 525 (25): 2540–2568. doi: 10.5897/AJB2006.000-5110
[22] BRUMFIELD R T, BEERLI P, NICKERSON D A, et al. The utility of single nucleotide polymorphisms in inferences of population history [J]. Trends Ecol Evol, 2003, 18(5): 249–256. doi: 10.1016/S0169-5347 (03)00018-1.
[23] MILLER M R, DUNHAM J P, AMORES A, et al. Rapid and cost- effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers [J]. Genome Res, 2007, 17(2): 240–248. doi: 10.1101/gr.5681207.
[24] PEGADARAJU V, NIPPER R, HULKE B, et al.sequencing of sunflower genome for SNP discovery using RAD (restriction site associated DNA) approach [J]. BMC Genom, 2013, 14(1): 556. doi: 10. 1186/1471-2164-14-556.
[25] MAO C L, ZHANG F L, LI X Q, et al. Genetic diversity ofbased on AFLP markers [J]. J Trop Subtrop Bot, 2020, 28(3): 271–276. doi: 10.11926/jtsb.4162.
毛常丽, 张凤良, 李小琴, 等. 琴叶风吹楠资源遗传多样性的AFLP分析[J]. 热带亚热带植物学报, 2020, 28(3): 271–276. doi: 10. 11926/jtsb.4162.
[26] DOYLE J J T, DOYLE J L. Isolation of plant DNA from fresh tissue [J]. Focus, 1990, 12(1): 13–15.
[27] DAVEY J W, BLAXTER M L. RADSeq: next-generation population genetics [J]. Brief Funct Genom, 2010, 9(5/6): 416–423. doi: 10.1093/ bfgp/elq031.
[28] ROCHETTE N C, RIVERA-COLÓN A G, CATCHEN J M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics [J]. Mol Ecol, 2019, 28(21): 4737–4754. doi: 10.1111/mec.15253.
[29] LISCHER H E L, EXCOFFIER L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs [J]. Bioinformatics, 2012, 28(2): 298–299. doi: 10.1093/bio informatics/btr642.
[30] EXCOFFIER L, LISCHER H E L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows [J]. Mol Ecol Resour, 2010, 10(3): 564–567.
[31] NEI M. Analysis of gene diversity in subdivided populations [J]. Proc Natl Acad Sci USA, 1973, 70(12): 3321–3323.
[32] OKSANEN J, BLANCHET F G, KINDT R, et al. Vegan: Community ecology package [CP/OL]. 2019. https://github.com/vegandevs/.
[33] PRITCHARD J K, STEPHENS M, DONNELLY P. Inference of population structure using multilocus genotype data [J]. Genetics, 2000, 155(2): 945–959.
[34] EARL D A, VONHOLDT B M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and imple- menting the Evanno method [J]. Conserv Genet Resour, 2012, 4(2): 359–361. doi: 10.1007/s12686-011-9548-7.
[35] JAKOBSSON M, ROSENBERG N A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multi- modality in analysis of population structure [J]. Bioinformatics, 2007, 23(14): 1801–1806. doi: 10.1093/bioinformatics/btm233.
[36] ROSENBERG N A. Distruct: A program for the graphical display of population structure [J]. Mol Ecol Notes, 2004, 4(1): 137–138. doi: 10. 1046/j.1471-8286.2003.00566.x.
[37] EVANNO G S, REGNAUT S J, GOUDET J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study [J]. Mol Ecol, 2005, 14(8): 2611–2620. doi: 10.1111/j.1365-294X. 2005.02553.x.
[38] ZHANG S S, KANG H M, YANG W Z. Population genetic analysis ofby reduced-representation sequencing technique [J]. Bull Bot Res, 2019, 39(6): 899–907. doi: 10.7525/j.issn.1673-5102. 2019.06.013.
张珊珊, 康洪梅, 杨文忠. 基于简化基因组技术的云南蓝果树群体遗传分析[J]. 植物研究, 2019, 39(6): 899–907. doi: 10.7525/j.issn. 1673-5102.2019.06.013.
[39] LI Q M, XU Z F, HE T H. A preliminary study on conservation genetics of endangered(Dipterocarpaceae) [J]. Acta Bot Sin, 2002, 44(2): 246–249. doi: 10.3321/j.issn:1672-9072. 2002.02.022.
李巧明, 许再富, 何田华. 濒危植物版纳青梅保护遗传学研究初报[J]. 植物学报, 2002, 44(2): 246–249. doi: 10.3321/j.issn:1672-9072. 2002.02.022.
[40] XU G B, LIANG Y, JIANG Y, et al. Genetic diversity and population structure of, an endangered species [J]. Biodiv Sci, 2013, 21(6): 723–731. doi: 10.3724/SP.J.1003.2013.09117.
徐刚标, 梁艳, 蒋燚, 等. 伯乐树种群遗传多样性及遗传结构[J]. 生物多样性, 2013, 21(6): 723–731. doi: 10.3724/SP.J.1003.2013. 09117.
[41] YAN S Y, ZHU P, GONG W, et al. Studies on genetic diversity ofcultivar germplasms in Sichuan based on RAD-SNPs analysis [J]. J Trop Subtrop Bot, 2019, 27(1): 19–28. doi: 10.11926/jtsb.3906.
闫思宇, 朱鹏, 龚伟, 等. 基于RAD-SNPs分析的四川核桃良种资源的遗传多样性研究[J]. 热带亚热带植物学报, 2019, 27(1): 19–28. doi: 10.11926/jtsb.3906.
[42] HAMRICK J L, GODT M J W, SHERMAN-BROYLES S L. Factors influencing levels of genetic diversity in woody plant species [J]. New For, 1992, 6(1): 95–124. doi: 10.1007/BF00120641.
[43] GE S. Review and prospect of genetic structure of plant population [M]// LI C S. Advances in Plant Science, Vol. 1. Beijing: Higher Education Press, 1977: 1–15.
葛颂. 植物群体遗传结构研究的回顾和展望[M]// 李承森. 植物科学进展, 第1卷. 北京: 高等教育出版社, 1977: 1–15.
[44] CHENG S L. Historical lessons from development of Hainan Island [J]. Mar Dev, 1987(1): 62–65.
成松林. 开发海南岛的历史教训[J]. 海洋开发与管理, 1987(1): 62–65.
[45] WRIGHT S. The interpretation of population structure by F-statistics with special regard to systems of mating [J]. Evolution, 1965, 19(3): 395–420. doi: 10.1111/j.1558-5646.1965.tb01731.x.
[46] GAMBA D, MUCHHALA N. Global patterns of population genetic differentiation in seed plants [J]. Mol Ecol, 2020, 29(18): 3413–3428. doi: 10.1111/mec.15575.
[47] FAEGRI K, VAN DER PIJL L. The Principles of Pollination Ecology [M]. 3rd ed. Oxford: Pergamon Press, 1979.
[48] NYBOM H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants [J]. Mol Ecol, 2004, 13(5): 1143–1155. doi: 10.1111/j.1365-294X.2004.02141.x.
[49] WANG C, MA X, REN M X, et al. Genetic diversity and population structure in the endangered tree(Dipterocarpaceae) on Hainan Island, China [J]. PLoS One, 2020, 15(11): e0241452. doi: 10.1371/journal.pone.0241452.
[50] YANG J, CAI L, LIU D T, et al. China’s conservation program on plant species with extremely small populations (PSESP): Progress and perspectives [J]. Biol Conserv, 2020, 244: 108535. doi: 10.1016/j.biocon. 2020.108535.
Genetic Diversity of: An Endangered Species with Extremely Small Populations
Cai Chaonan1,2, Hou Qinxi3, CI Xiuqin1,4, Xiao Jianhua1,5, Zhang Canyu1,5, LI Jie1,4*
(1.Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences,Kunming 650223, China; 2. School of Advanced Study, Taizhou University, Taizhou 318000, Zhejiang, China; 3. Sichuan Academy of Giant Panda, Chengdu 610081, China; 4. Core Botanical Gardens, Chinese Academy of Sciences,Mengla 666303, Yunnan, China; 5. University of Chinese Academy of Sciences,Beijing 100049, China)
In order to explore the causes of the endangered species, the genetic diversity and population structure were analyzed by using restriction-site associated DNA sequencing (RAD-seq) for developing single nucleotide polymorphisms (SNPs). The results showed that the genetic diversity ofwas low (=0.167) and the genetic differentiation between populations was significant (F=0.120). Structure analysis showed that the optimal clustering value of the population was 2, but the genetic structure of some populations was mixed, which was consistent with the results of Mantel correlation test showing no correlation between genetic distance and geographic distance (=0.733,<0.075). Therefore, low regeneration ability and excessive disturbance of human activities might be the main reason for endangered status of. It was recommended to strengthenconservation of populations with high genetic diversity, such as BWL and YGL, and strengthenorconservation of populations with severe habitat damage, such as EXL and DLS, to increase gene exchange among populations. At the same time construct the core germplasm of this species to prevent the aggravation of genetic resource loss.
; RAD-seq; Genetic diversity; Extremely small population; Genetic structure
10.11926/jtsb.4364
2020-12-21
2021-02-24
科技部科技基础资源调查专项(2017FY100100)资助
This work was supported bythe Program for Science and Technology Basic Resources Investigation of Ministry of Science and Technology (Grant No. 2017FY100100).
蔡超男(1992~ ),女,博士,讲师,研究方向为植物系统发育与濒危物种的保护遗传学。E-mail: caichaonan@xtbg.ac.cn
. E-mail: jieli@xtbg.ac.cn
猜你喜欢
今日农业(2022年15期)2022-09-20 06:54:16
河北科技师范学院学报(2022年2期)2022-08-26 08:55:24
浙江中医药大学学报(2021年6期)2021-07-12 03:05:14
World Journal of Integrated Traditional and Western Medicine(2019年3期)2019-12-21 14:44:45
草地学报(2018年5期)2018-11-07 02:25:00
红土地(2018年7期)2018-09-26 03:07:38
小学生作文(中高年级适用)(2018年4期)2018-05-14 06:30:19
南方周末(2018-05-03)2018-05-03 17:02:13
音乐天地(音乐创作版)(2017年10期)2018-01-25 22:48:16
当代畜禽养殖业(2014年10期)2014-02-27 07:59:49
