
次亚磷酸含量测定新方法的研究
2021-10-12 03:57周骏宏陈杭馨陈生悦万宗静
无机盐工业 2021年10期
郑 娜,周骏宏,陈杭馨,陈生悦,万宗静,盛 余
(黔南民族师范学院化学化工学院,贵州都匀558000)
磷在生产生活中的应用非常广泛,对国民经济有着极其重要的作用。磷含量作为磷化工行业的常规测定项目,其测定方法分为化学分析法和仪器分析法两种。其中,仪器分析法中实验室最常见的是分光光度法[1],主要又分为磷钒钼黄法和磷钼蓝法及离子缔合物法,在高端分析领域仪器分析法还有离子色谱法[2]和ICP-AES法[3-5];而化学分析方法则主要是喹钼柠酮重量法和容量法,两种都是实验室常用的测磷含量的方法。其中重量法准确度极高,并且测量范围广(大约为30~130 mg/L),因而被定为仲裁法。
分光光度法测量样品中磷含量,一般采用喹钼柠酮法、磷钒钼黄法、磷钼蓝法,都是基于形成磷钼杂多酸的测定方法,其原理是正磷酸盐在酸性条件下与钼酸盐等定量反应形成磷钼杂多酸,通过测定磷钼杂多酸的含量算出磷含量[6]。一般需要消解或灰化法处理样品[7-8]。但在喹钼柠酮重量法和容量法中,样品的预处理却鲜有研究,而其他容量法对次亚磷酸的测定一般采用的是氧化还原滴定法,但存在滴定终点不灵敏导致测定误差较大的问题。而重量法测样品的磷含量虽然具有准确性高的优点,但由于重量法目前少有预处理方法,所以只适用于正磷酸根的测定,对次亚磷酸完全不适用,所以不能直接采用重量法进行次亚磷酸的测定。
重量法测定磷组分含量具有准确度高的优点(尤其是对高含量样品),但由于其存在不适用于低价磷的缺陷,即喹钼柠酮重量法无法直接测量次亚磷酸及盐的问题,笔者提出一种先用合适的氧化剂将次亚磷酸全部氧化为正磷酸,再用重量法测定的思路并进行探究。此法可适用于测定含磷量较高且含有以非正磷酸形式存在的磷样品的总磷含量的准确测定,并且此新方法有准确度较高、适测范围广、可广泛应用的特点。
1 实验方法
1.1 主要仪器和试剂
仪器:分析天平、循环水多用真空泵、电热恒温鼓风干燥箱、G4玻璃坩埚、玻璃漏斗。
试剂:浓硝酸、0.1 mol/L硝酸铈铵溶液、喹钼柠酮沉淀剂。
喹钼柠酮沉淀剂的配制:1)称取70 g钼酸钠溶解于150 mL水中,此溶液为溶液A;2)称取60 g柠檬酸溶解于150 mL水和85 mL硝酸的混合溶液中,此溶液为溶液B;3)在搅拌下将溶液A倒入溶液B中,此溶液为溶液C;4)在100 mL水中加入35 mL硝酸,再加入5 mL喹啉,此溶液为溶液D;5)将溶液D倒入溶液C中,混匀。放置12 h后,用玻璃砂坩埚过滤,再加入280 mL丙酮,用水稀释至1 000 mL混匀,贮存于聚乙烯瓶中。
1.2 实验步骤
1.2.1 氧化
取1.000 0 g(精确到0.000 1 g)试样,置于100 mL小烧杯溶解,移入500 mL容量瓶,用水稀释至刻度,摇匀,制得待测溶液。取待测溶液10 mL置于250 mL烧杯中,加入纯水至100 mL,加入10 mL浓硝酸,再加入15 mL硝酸铈铵溶液。在烧杯中放入玻璃珠或沸石,并盖上表面皿,置于电热板上加热,使溶液沸腾15 min以上,确保溶液中次亚磷酸全部充分氧化,并且使过量的硝酸铈铵分解。
1.2.2 沉淀
在氧化好的样品溶液中加入5 mL浓硝酸,再加入50 mL喹钼柠酮试剂。将其置于75℃水浴锅中加热数分钟,停止加热。取下烧杯,冷却至室温。用预先恒重好的玻璃坩埚抽滤,将沉淀转移到玻璃坩埚中,置于250℃(±5℃)鼓风干燥箱中干燥至恒重,然后置于干燥器中冷却20~30 min至室温,称量。
1.3 测定结果的计算
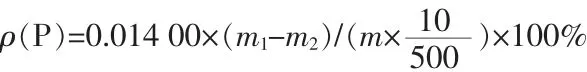
其中:0.014 00为磷钼酸喹啉摩尔质量换算为磷摩尔质量的系数;m1为坩埚与沉淀的质量,g;m2为坩埚的质量,g;m为称取的样品质量,g。
2 结果与讨论
2.1 磷钼酸喹啉沉淀形成条件的探究
磷钼酸喹啉是一种大分子的、溶解度很小的难溶盐,该沉淀在硝酸酸性介质中生成,利用硝酸的氧化性来保证磷和沉淀剂中的钼均以高价状态存在。在酸性介质中,正磷酸根与喹钼柠酮沉淀剂反应生成黄色磷钼酸喹啉沉淀,经过滤、洗涤、干燥和称量所得沉淀,根据沉淀质量换算出五氧化二磷的含量。喹钼柠酮只能沉淀正磷酸,而对于待测样品中的亚磷酸、次磷酸的成分,喹钼柠酮无法测定。
为了验证喹钼柠酮与次磷酸、亚磷酸(根)等不同形态的磷是否正常响应而形成沉淀,分别用正磷酸、次磷酸、亚磷酸进行对照实验,结果如表1所示。
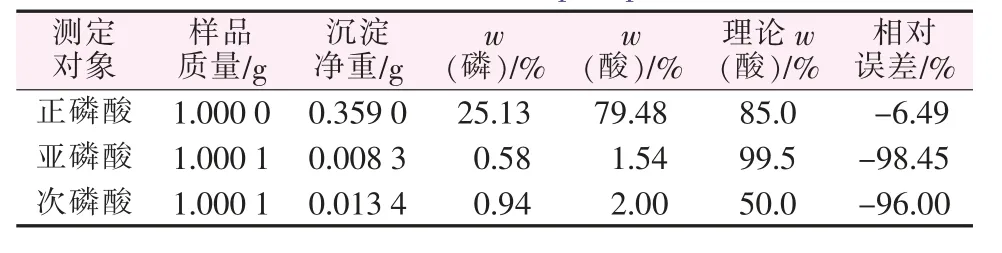
表1 原方法对不同形态的磷的测定Table 1 The original method for determination of different forms of phosphorus
可见,在次亚磷酸含量的测定中,原始的重量法根本无法得到沉淀,几乎无法测出其含量,说明喹钼柠酮不与次亚磷酸响应,原重量法不适用于测量次亚磷酸及盐的磷含量,意味着采用传统的重量法不可能测出次亚磷酸等低价态的磷的含量。这也是至今未看到有重量法运用在低价态磷测定上的报道的原因。沉淀反应方程式如下:
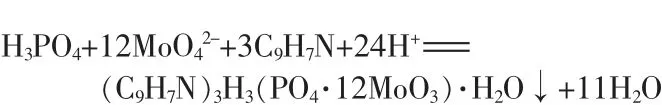
喹钼柠酮沉淀剂仅能与五价磷作用,即使在实验条件的强硝酸介质加热情况下也一样,而与三价、一价磷不反应,更不能形成沉淀。所以,要想采用喹钼柠酮重量法测定低价形态的磷,只能先氧化低价磷将其完全转化为五价磷,再进行测定。
2.2 氧化剂的筛选
本实验在测定磷样时,在使用沉淀剂喹钼柠酮前使用氧化剂将亚磷酸、次磷酸全部氧化为正磷酸,再与沉淀剂喹钼柠酮生成沉淀磷钼酸喹啉(黄色)。发生的反应如下:
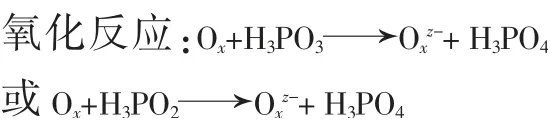
此时要准确测定磷含量就需选取合适的氧化剂,本实验氧化剂的选择原则是:1)能存在于酸性介质中完全氧化次亚磷酸;2)不影响后续测定。
首先,针对第一条,即氧化剂必须在本实验条件下具有氧化能力,开展筛选实验。结果发现,当分别加入双氧水、高锰酸钾、硝酸铈铵3种试剂与次亚磷酸配制的待测液氧化后再加入沉淀剂喹钼柠酮,由不能沉淀变为均有明显沉淀。上述试验初步说明3种氧化剂均可将次亚磷酸氧化为正磷酸,具有本实验条件下的氧化能力。但是进一步的实验发现,过量的H2O2又会与沉淀剂发生反应,导致结果偏低或者浪费沉淀剂[9]。因此双氧水并非理想的次亚磷酸氧化剂。高锰酸钾能够有效氧化次亚磷酸,将其全部氧化为正磷酸根,且过量的高锰酸钾对于喹钼柠酮没有影响;但高锰酸钾作为本实验的氧化剂的用量难以掌握,增加操作难度,如果控制不好会产生较大误差。
相比高锰酸钾和双氧水,硝酸铈铵用于氧化次磷酸、亚磷酸氧化效果理想、操作更为简便易行,故对于硝酸铈铵笔者做了更深入的探究。
2.3 氧化条件探究
为了掌握硝酸铈铵氧化次亚磷酸并与喹钼柠酮沉淀测出磷含量的最适宜实验条件,需要研究反应条件、氧化剂用量等对测定结果的影响。由于喹钼柠酮重量法在沉淀过程中本身需加热处理,而硝酸铈铵亦需在加热条件下体现其强氧化性,由此确定本实验条件需加热,故对此不再深入研究。因此本实验重点研究硝酸铈铵的用量的影响。
2.3.1 硝酸铈铵的用量探究
硝酸铈铵是一种氧化剂,在本实验条件下可以氧化次亚磷酸根,形成五价磷化合物,反应化学方程式分别为:
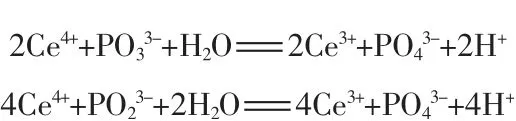
根据计算,使用本方法的硝酸铈铵的理论用量:氧化亚磷酸需要0.1 mol/L的硝酸铈铵7.3 mL,氧化次磷酸需要0.1 mol/L的硝酸铈铵12.12 mL。
称取1.000 0 g亚磷酸样品,加入不同量、浓度为0.1 mol/L的硝酸铈铵,对硝酸铈铵的用量进行探究,结果见图1。
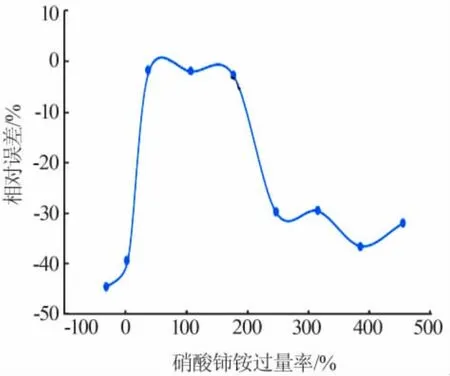
图1 加入不同量硝酸铈铵氧化亚磷酸Fig.1 Adding different amounts of cerium ammonium nitrate to oxidize phosphorous acid
从图1可知,硝酸铈铵过量率为36.99%~173.97%时相对误差很小,超出此范围都会导致较大的测定误差,导致测量结果偏小。氧化剂用量不足会使次亚磷酸的氧化反应不完全而导致测量结果偏小。但氧化剂过量也会导致测得值变小并且过量越多偏离越大。对此,采用正磷酸为对象,探究加入大量的硝酸铈铵对正磷酸的测定是否会造成影响。结果发现,在含大量正磷酸的待测液中加入大量硝酸铈铵时会有溶液略微浑浊的现象。但同时也发现如果加入硝酸铈铵的量不大,对正磷酸测定的影响也在合理范围内,其结果见表2。
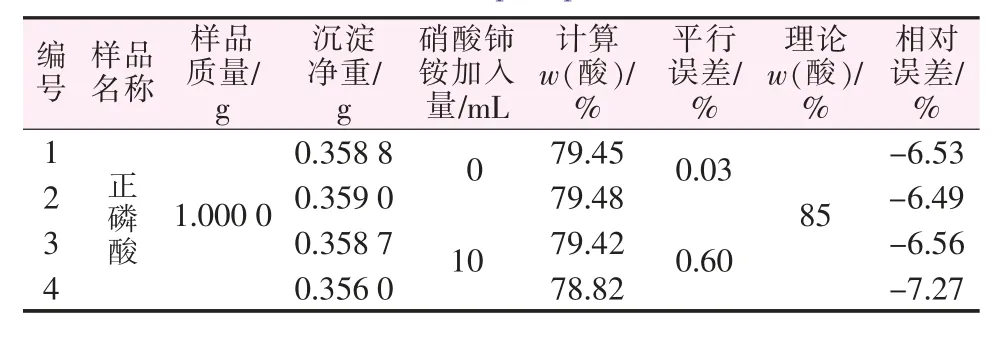
表2 硝酸铈铵对正磷酸影响的探究Table 2 Exploration of the effect of cerium ammonium nitrate on orthophosphoric acid
综上可知:对于未知其组成的样品测定,硝酸铈铵(0.1 mol/L)的实际用量应高于所计算的理论用量12.12 mL,加之硝酸铈铵过多会对正磷酸有影响,过量的硝酸铈铵会导致测定结果偏低,因此硝酸铈铵也不宜过多。笔者认为硝酸铈铵的用量在15~20 mL较合适。但实际测定情况往往不知道样品的磷含量是多少,所以无法确定过量的合适范围,对此还需要有进一步的解决方案。
2.3.2 异常情况及消除
由2.3.1节可知加入过量的硝酸铈铵会导致测定结果偏小,对此笔者经实验发现过量的硝酸铈铵与沉淀剂喹钼柠酮混合后会出现有少量沉淀的现象:不加待测液,于250 mL烧杯中加入100 mL去离子水,加入100 mL硝酸铈铵,加热至沸,再加入50 mL沉淀剂。静置后烧杯底部有少量黄色沉淀。
理论上过量的硝酸铈铵可能会因为产生其他沉淀而导致结果偏大,也可能会因为消耗沉淀剂而使结果偏小。结合实验数据,总体而言其结果是沉淀剂被消耗而导致结果偏小。必须在加入喹钼柠酮之前将过量的硝酸铈铵除去。
硝酸铈铵的氧化作用需要加热,实验发现如果在电热板上沸腾后再加热,过量的硝酸铈铵会分解,故拟以加热分解的方式除过量的硝酸铈铵。
若在过量的硝酸铈铵分解完全时加入少量硝酸、50 mL喹钼柠酮,则不会再有沉淀产生干扰实验。若加热时间不够长,过量的硝酸铈铵未分解完全,则会因为与喹钼柠酮反应而使结果极其不稳定,影响其测定。实验结果见表3、表4。
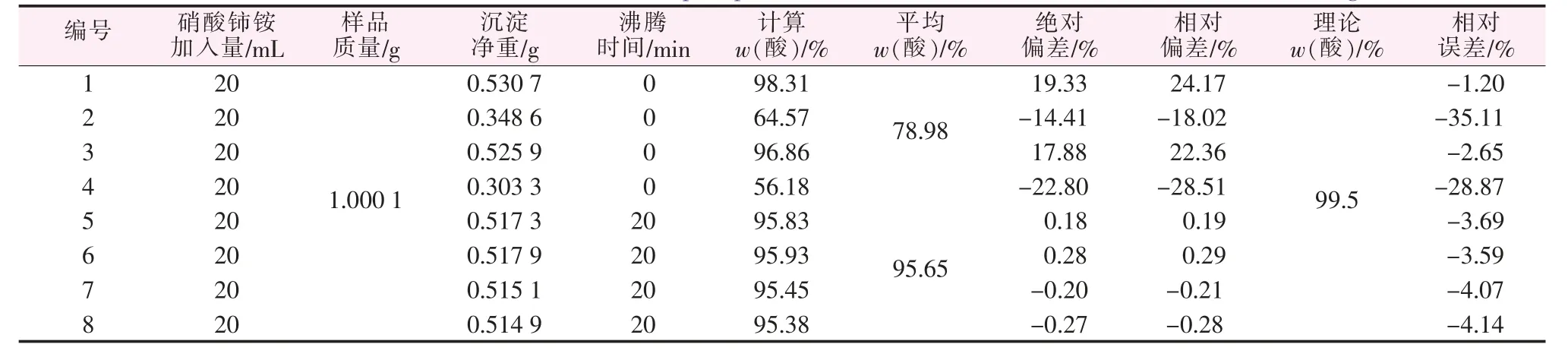
表3 亚磷酸与硝酸铈铵混合液加热后测定Table 3 Determination of the mixture of phosphorous acid and cerium ammonium nitrate after heating

表4 次磷酸与硝酸铈铵混合液加热后测定Table 4 Determination of the mixture of hypophosphorous acid and cerium ammonium nitrate after heating
如表3、表4所示,若仅仅将待测液与硝酸铈铵的混合溶液加热至刚好沸腾,如表3编号1~4,测定值极其不稳定,误差很大,但沸腾20 min可将过量的硝酸铈铵除尽,再加入硝酸与沉淀剂,则测定值偏差更小。由此可知,溶液中过量的硝酸铈铵必须除尽,且方法为加热沸腾20 min以上使其分解。
3 最适宜实验条件下对实际样品的测定
为验证本实验方法的可靠性,选用不同组成的混合磷样品、组成未知的实际磷样品以及标准物按照本实验方法检验。
3.1 混合磷样的测定
取正磷酸、亚磷酸、次磷酸各1.000 1 g分别制成500 mL水溶液,再根据正磷酸、亚磷酸、次磷酸不同的配比配制样品,加入0.1 mol/L的硝酸铈铵氧化后再测定,其结果见表5~7。由表5~7可以看出,不加入氧化剂硝酸铈铵时,因为此时仅能测出混合磷样中正磷酸形态的磷,次磷酸、亚磷酸形态的磷没有响应,不会形成沉淀,故混合磷样的测定结果比真实值偏小很多。而当加入氧化剂后,沉淀包含了次亚磷酸形态的磷,才能准确反映出全部磷化合物的含量,并由此可得出次亚磷酸含量。为了进一步验证本法,再取两种未知组成的含磷样品配制待测液,用本实验方法测其磷含量,结果见表8。
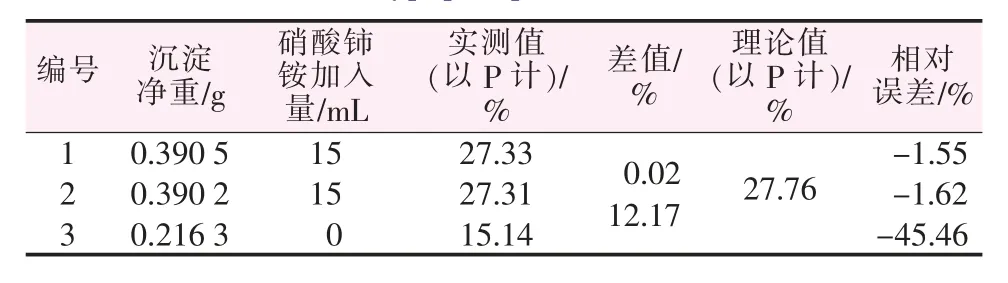
表5 含正磷酸60%、亚磷酸20%、次磷酸20%的混合样测定结果Table 5 Determination results of mixed sample of orthophosphoric acid 60%,phosphorous acid 20%and hypophosphorous acid 20%
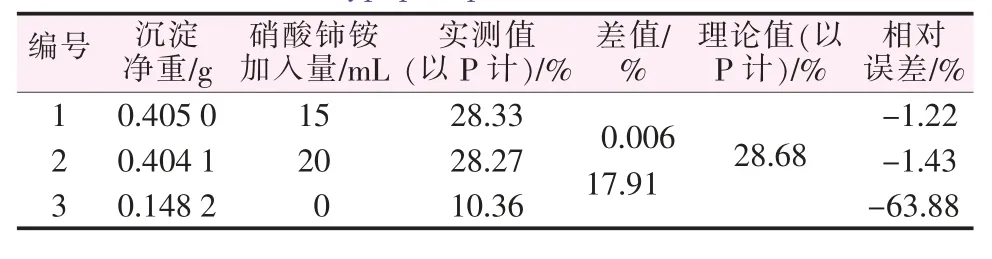
表7 含正磷酸40%、亚磷酸30%、次磷酸30%的混合样测定结果Table 7 Determination results of mixed sample of orthophosphoric acid 40%,phosphorous acid 30%,and hypophosphorous acid 30%

表8 亚磷酸亚铁等未知样品的测定Table 8 Determination of unknown samples such as ferrous phosphite
硝酸铈铵对正磷酸、亚磷酸、次磷酸含量不同试样的氧化效果都很好且结果稳定,且由表8可知对于亚磷酸亚铁等次亚磷酸盐的样品同样适用,有实际应用的意义。
3.2 准确度试验
选取次磷酸钙为标准物,取1.000 1 g用本法测定,其结果见表9。由表9可知,本法相对偏差、相对误差均<1%。
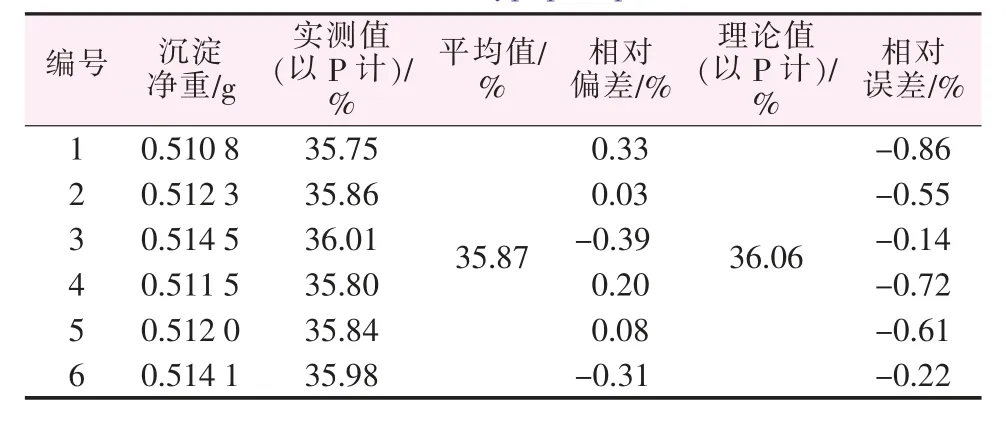
表9 对次磷酸钙中磷含量的测定Table 9 Determination of phosphorus content in calcium hypophosphite
4 结论
1)使用传统的喹钼柠酮重量法无法测定低价磷组分的含量,但采用合适的氧化剂先氧化低价磷转变为五价磷,则能够继续运用喹钼柠酮重量法测定,从而解决传统重量法无法准确测量含亚磷酸、次磷酸及其盐等样品的问题。
2)初步筛选出的氧化剂的选择:高锰酸钾、双氧水、硝酸铈铵3种氧化剂都能完全氧化次亚磷酸,但高锰酸钾和双氧水都有氧化终点无法确定的问题,且双氧水会影响喹钼柠酮沉淀剂,仅有硝酸铈铵氧化剂可适用于本法。
3)实验结果表明,使用0.1 mol/L硝酸铈铵氧化某一待测溶液中的次磷酸和亚磷酸用量为15~20 mL为宜。无法判断过量程度的情况下,可以加入较多的硝酸铈铵,多余的硝酸铈铵采用加热至沸腾20 min以上的办法可将过量的硝酸铈铵除尽。
4)此法对于同时含有次亚磷酸、正磷酸的样品,可以分别测出样品中含有的正磷酸含量、总磷含量、次亚磷酸含量,有助于更好了解具有多形态、多组成的含磷物质的性质,更有实际应用的价值。
猜你喜欢
热带作物学报(2021年10期)2021-12-08
中学生数理化·高一版(2020年11期)2020-12-14
中国化工贸易·下旬刊(2020年3期)2020-10-21
河北工业大学学报(2019年3期)2019-09-10
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
中学化学(2019年2期)2019-07-08
浙江农业学报(2017年1期)2017-05-17
中国资源综合利用(2016年6期)2016-01-22
科技创新导报(2014年20期)2014-11-10
