
基于曼德尔h/k 统计法评价16 家实验室盲样考核结果
2021-10-11 09:30:18何虹朱荣丁建东卢日刚
化学分析计量 2021年9期
何虹,朱荣,丁建东,卢日刚
(广西壮族自治区食品药品检验所,南宁 530021)
为了验证实验室检测能力,确保检测结果的准确性和可比性,定期开展实验室间比对活动,能直接反映实验室的仪器设备和技术素质的实际水平和出具报告的能力[1-2]。本次诺氟沙星含量测定的盲样考核所采用的检验方法,参考药品原料药诺氟沙星的现行标准[3],采用目前药品检验常用的高效液相色谱法测定[4]。本次盲样考核用曼德尔h/k统计替代通常的离群值检验方法,避免冒失地剔除不一致的数据导致方法精密度的误判,帮助考察实验室检测人员(QC)对于高效液相色谱法的检测能力,以及监控实验室的持续检测能力,有助于识别实验室的问题并采取纠正措施,提高质量管理水平。
1 样品与方法
1.1 盲样考核样品
本次盲样考核样品为诺氟沙星样品,由南宁金马制药厂(有限公司)生产,样品呈类白色粉末,包装于棕色瓶内。经均匀性和稳定性评价合格后的样品,作为盲样考核用样品发送给参加本次盲样考核的16 家实验室[5]。
为了更加客观、公正地评价参加实验室结果及防止实验室间数据串通,本次盲样考核配送给每个实验室1 个工作对照品、1 个样品,以及2 个干扰样品(准备3 个干扰样品,每个实验室随机配送2 个,干扰样品不参与结果评判)。
1.2 样品测定方法
1.2.1 仪器工作条件
色谱柱:十八烷基硅烷键合硅胶为填充剂;流动相:0.025 mol/L 磷酸溶液(用三乙胺调pH 值至3.0)-乙腈(87∶13);检测波长:278 nm;流量:1.0 mL/min;进样体积:20 μL。
1.2.2 样品处理
(1)供试品溶液的制备。取细粉样品100 mg,精密称定,置于200 mL 容量瓶中,加0.1 mol/L盐酸溶液4 mL 溶解,用水稀释至标线,摇匀,精密量取续滤液5 mL,置于50 mL 容量瓶中,用流动相稀释至标线,摇匀,作为供试品溶液。
(2)对照品溶液的制备。取诺氟沙星工作对照品25 mg,精密称定,加适量0.1 mol/L 盐酸溶液溶解,同法操作,用流动相稀释成诺氟沙星质量浓度为25 mg/mL 对照品溶液。
1.2.3 样品测定
将仪器调至最佳状态,取诺氟沙星供试品溶液和对照品溶液各20 μL 注入液相色谱仪,记录诺氟沙星色谱峰的保留时间和峰面积,以保留时间作为定性依据,以色谱峰面积作为定量依据,按外标法-单点校正法计算诺氟沙星样品的含量。诺氟沙星样品的含量按照公式(1)计算:

1.2.4 样品结果
以百分含量报告测定结果(按照对照品计算,不扣除水分),保留小数点后2 位,并将两次测定的结果、平均值以及相对标准偏差填入结果报告单中。
1.3 评价方法
本次盲样考核采用安德森-达林(AD)检验和曼德尔(Mandel's)h/k检验进行统计分析和评价。
1.3.1 安德森-达林统计检验
安德森-达林(AD)统计检验可用于度量数据服从特定概率分布的程度。AD 统计检验按照公式(2)计算。

1.3.2 曼德尔h/k统计量检验
曼德尔h/k统计量检验用来评定分析数据的一致性,该统计属于单水平下实施的单因素方差分析。h值是实验室间一致性的统计量,即水平下某参加实验室与水平总体之差的度量,反映单个实验室实验结果的均值与所有实验室结果均值的偏差。k值是把单个实验室内的重复性偏差与所有参与盲样考核实验室的平均重复性偏差进行对比,反映单个实验室内各实验结果与其均值相比,其它实验室的偏差。h-crit 临界值和k-crit 临界值分别按公式(3)和公式(4)计算[6]:

采用曼德尔h/k统计量检验,既可以检验实验室间测试结果的准确度,又可以检验实验室内测试结果的精密度。一般情况下,某个实验室的h值越大,表明其测试结果准确度越低。当h值不大于5%的临界值时,认为该值为可信值;当h值大于5%的临界值且不大于1%的临界值时,认为该值为歧离值;当h值大于1%的临界值时,认为该值为离群值。某个实验室的k值越大,表明其实验室测试结果的精密度越低。当k值不大于5%的临界值时,认为该值为可信值,当k值大于5%的临界值且不大于1%的临界值时,认为该值为歧离值;当k值大于1%的临界值时,认为该值为离群值。
2 结果与分析
2.1 参加对象
本次盲样考核的对象为广西壮族自治区内9 个市的16 家实验室,其中9 家为自治区内各市级的药品检验实验室,其余7 家为自治区内有GMP 认证的药品生产企业QC 实验室。样品发放前,由广西壮族自治区食品药品检验所负责样品均匀性和稳定性评价。
2.2 盲样考核结果
参加盲样考核的16 个实验室(代码为i=A01、A02、A03、A04、A05、A06、A07、A08、A09、A10、A11、A12、A13、A14、A15、A16),按照1.2 检验方法对盲样中诺氟沙星的含量进行测定,每个样品进行2 次独立测定(编号为j=1、2),16 家参加盲样考核实验室的检测结果见表1。
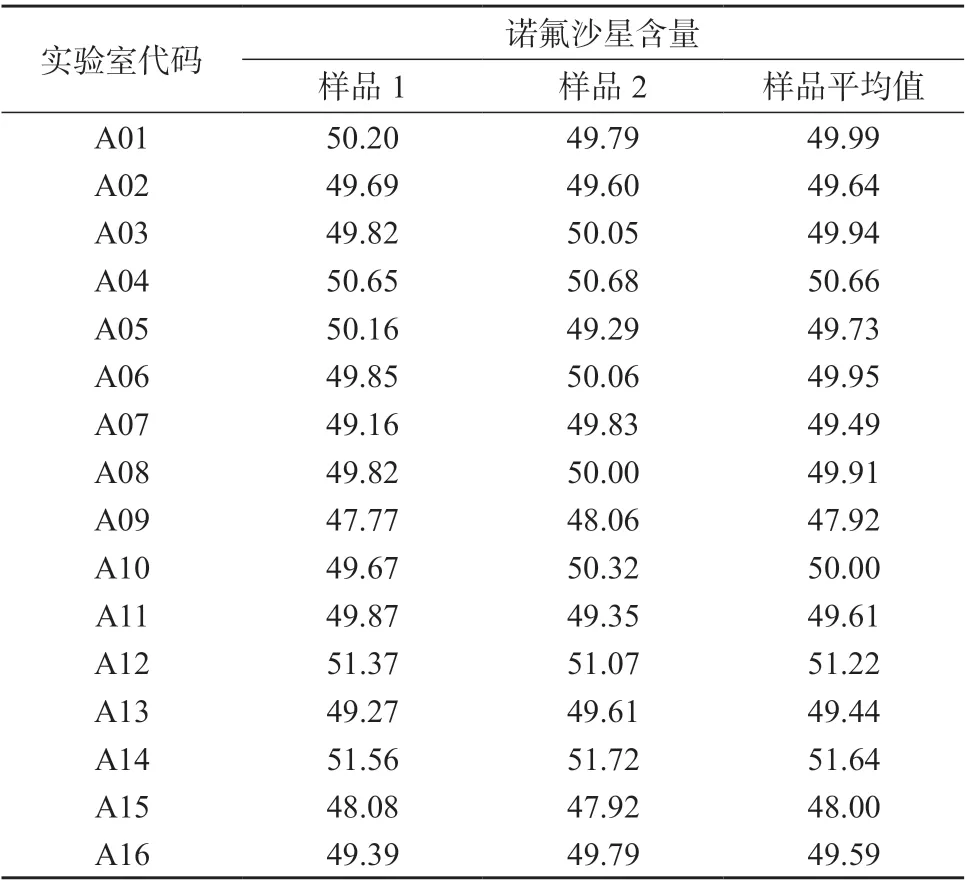
表1 16 家参加盲样考核实验室的检测结果 %
2.3 盲样考核结果数据统计分析
因参加实验室的数量未能满足稳健统计[7]的要求,故剔除每家实验室的干扰样品值后,固定检测样品分别采用安德森-达林检验、曼德尔h/k统计检验,对16 家参加实验室的盲样考核结果进行单因素方差统计分析。
2.3.1 测定结果的安德森-达林统计检验
根据表1 中16 家参加盲样考核实验室的检测结果数据,采用安德森-达林检验统计分析,运用公式(2)计算出16 家实验室结果数据的安德森-达林统计检验量见表2。

表2 16 家实验室盲样考核结果AD 统计量

2.3.2 测定结果的曼德尔h/k统计量检验
根据表1 中检测结果数据,按公式(3)和公式(4),采用曼德尔h/k检验统计分析,计算出16 家实验室的曼德尔h统计量和k统计量见表3。

表3 实验室间检测结果的曼德尔h/k 统计检验表
在95%的概率下,分别绘制各实验室的曼德尔h值分析柱状图(图1)和k值分析柱状图(图2)。

图1 水平样品下的实验室间检测结果h 统计检验量柱状图

图2 水平样品下的实验室间检测结果k 统计检验量柱状图
结合表3 数据,从h值统计图中(图1)可知,在曼德尔h的统计结果中,实验室A09、A15 及A14的结果分别为-1.990、-1.900、1.951,属于歧离值,其余13 家结果为正常值。由k值统计图(图2)可知,在曼德尔k的统计结果中,实验室A05 的结果分别为2.115,属于歧离值,其余14 家结果为正常值。若仅根据实验室间曼德尔h/k检验统计量判定实验室结果可疑,容易导致误判的可能,需要进一步分析。经查看上述4 家实验室盲样考核原始数据的记录,表明不存在样品前处理、程序、誊抄等方面的失误。随后征询了A05、A09、A14 及A15 实验室的意见,这4 家实验室均认为是严格遵守规范进行操作。曼德尔h和k检验中的歧离值属于随机误差,因此16 家实验室的盲样考核结果满意。
3 技术分析与建议
3.1 技术分析
本次盲样考核共发放3 个不同浓度的样品,除1 个固定检测样品(A)外,随机发放2 个干扰样品。采用曼德尔h/k检验统计分析方法,对16 家实验室的所有测定数据进行分析,分别计算每个浓度样品参加实验室上报数据的k值,发现共有7 家实验室收到相同的3 个不同浓度样品(A、C、D)。综合这7 家实验的k值数据,绘制出3 个浓度样品的k值分析图(图3)并进行比较。分析发现A04 实验室3 个不同浓度k值均接近0,表明该实验室提交的检测数据可信度差[14]。从图3 分析图中可更直观的显示7 家实验室3 个不同浓度的检测情况,疑存检验人员修改数据的情况发生。该问题应引起A04 实验室管理者的高度注意,应对人、机、料、法、环等因素进行核查[15]。

图3 3 个浓度样品数据的k 值分析图
本次盲样考核的16 家实验室有9 家为本区域内的药品检验实验室,其余7 家为药品生产企业QC 实验室,从原始数据(表1)分析,采用安德森-达林检验和曼德尔h/k检验的各参加实验室的测试结果进行统计分析,简单、直观、有效,且利于比较,及时发现实验室存在的不足,了解与其它实验室的差距,便于进行下一步改进[16]。
3.2 改进建议
基于曼德尔h/k统计法评价这次的盲样考核结果,发现本次参加的A04 实验室3 个不同浓度k值均接近0,可再组织一次同类型多水平浓度盲样考核,追踪该实验室提交的数据,若再次发现A04实验室出现此类情况,可以表明该实验室检验人员有修改数据的情况存在。实验室盲样考核使用曼德尔h/k统计,可及时发现问题,真正起到发挥质量管理工具的作用。
4 总结
采用曼德尔h/k检验方法评估盲样考核结果的标准差与准确度后,各个实验室技术表现的优劣和误差可以被确定。每一个参加者可以了解其结果在本次盲样考核计划中所处的水平,对于各自存在的问题可以进一步查证原因,及时改正。今后应继续加强药品检测人员的业务综合技能的培训,积极组织多种形式的内外部能力验证,提升QC 检验人员的药品检测能力水平。建议各实验室在检测过程中,确保一切检测活动可控、可追溯、可重现,保证实验室质量控制措施的落实。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:23:00
昆明医科大学学报(2021年8期)2021-08-13 08:59:14
食品安全导刊·下旬刊(2020年6期)2020-09-10 01:38:31
东西南北(2020年1期)2020-03-05 00:38:54
绿色科技(2019年18期)2019-11-22 14:33:43
食品安全导刊(2019年7期)2019-08-06 17:36:42
妇女生活(2019年1期)2019-01-17 02:14:28
保健与生活(2019年1期)2019-01-13 13:54:39
看世界(2019年26期)2019-01-10 01:33:03
广东药科大学学报(2016年3期)2016-07-27 00:54:57
