
连续流动分析法测定杏仁中氰化物
2021-10-11 09:30:18胡馨月吴永强赖晋锋林莉刘娅
化学分析计量 2021年9期
胡馨月,吴永强,赖晋锋,林莉,刘娅
(1.泸州市疾病预防控制中心,四川泸州 646000; 2.西南医科大学公共卫生学院,四川泸州 646000)
氰化物是含氰基的一类化合物的总称,是广泛存在于自然界的剧毒物质,可抑制组织细胞生物氧化酶活性,阻滞三羧酸循环,造成“细胞内窒息”。生活中氰化物中毒以口服为主(如误食含氰果仁)。GB 5009.36—2016 《食品安全国家标准 食品中氰化物的测定》中提及的定量检测方法主要有分光光度法及气相色谱法,但并未包含杏仁中的氰化物检测。在食品监测工作中,杏仁中氰化物的检测一般采用气相色谱法,但该方法精密度偏差过大且偏差方向难控,方法中因试剂等影响造成批次间电子俘获(ECD)响应重复性差。目前连续流动仪测定氰化物的方法广泛应用于职业卫生检测、环境检测及食品检测等众多领域[1-4]。韦小烨等[5]建立的鲜木薯中氰化物的流动注射测定法线性范围宽、重复性好,对于油脂含量较少的样品,可以直接水解提取,取上清液过滤后进行分析。然而,对于含有大量油脂的样品,如杏仁,如何在前处理过程中除去干扰,同时做到高回收率及高精密度等结果的测定方法尚未见报道。结合食品监测方法及部分研究成果[6-8],研究人员建立了连续流动分析仪测定氰化物的方法,杏仁样品经前处理后,仪器密闭蒸馏出氢氰酸,与氯胺T 反应生成氯化氰,以异烟酸-巴比妥酸作染料,代替异烟酸-吡唑啉酮规避苯甲醛与吡唑啉酮反应产生白色混浊的干扰[9-10],生成物于600 nm处比色定量[11-13]。笔者建立连续流动分析法测定杏仁中的氰化物,采用正己烷萃取除去油脂,滤膜过滤除去固体颗粒,拓展了杏仁中氰化物检测的方向。
1 实验部分
1.1 主要仪器与试剂
连续流动分析仪:SKALAR SAN5000 型,带有四针自动进样器及循环冷却水系统,荷兰SKALAR公司。
气相色谱仪:安捷伦7890A,带有CTC 顶空进样器及ECD 检测器,美国安捷伦科技公司。
超声清洗仪;KQ2200DB 型数控,昆山超声仪器有限公司。
电子分析天平:AL204-IC 型,感量为0.001 g,瑞士梅特勒-托利多公司。
低温冷冻离心机;SIGMA2-16K 型,德国希格玛公司。
氰化物标准溶液:50 mg/L,编号为GBW(E)080115,中国计量科学研究院。
水相针式滤器(聚醚砜):13 mm×0.45 μm。
氢氧化钠、邻苯二甲酸氢钾、氯胺T、1,3-二甲基巴比妥酸、异烟酸、正己烷:分析纯,国药集团化学试剂有限公司。
盐酸:分析纯,天津市大茂化学试剂厂。
月桂醇聚氧乙烯醚(Brij35):CAS 号为9002-92-0,荷兰SKALAR 公司。
实验用水为超纯水。
1.2 溶液配制
pH5.2 的缓冲溶液:称取氢氧化钠2.3 g 溶解于500 mL 超纯水中,加入20.5 g 邻苯二甲酸氢钾,加超纯水至975 mL 后,分别用1 mol/L 的盐酸和氢氧化钠溶液调节pH 至5.2,定容至1 L 后加入1 mL 月桂醇聚氧乙烯醚,充分溶解后过滤,于棕色瓶中冷藏,待用。
氯氨T 溶液:称取氯胺T 2.0 g,用超纯水定容至1 000 mL,过滤后使用。
显色剂:称取氢氧化钠7 g 于约500 mL 超纯水中,再加入1,3-二甲基巴比妥酸16.8 g、异烟酸13.6 g,加入970 mL 超纯水溶解,用1 mol/L 的盐酸和氢氧化钠溶液调节pH 至5.2。
氰化物系列标准工作溶液:吸取2 mL 氰化物标准溶液于50 mL 容量瓶中,用超纯水定容至标线,得2.0 mg/L 氰化物标准溶液,再分别吸取0、0.25、0.50、1.0、1.5、2.5、5.0、12.5 mL 上述2.0 mg/L氰化物标准溶液于比色管中,用超纯水定容至25.0 mL,配制成氰化物质量浓度分别为0.02、0.04、0.08、0.12、0.20、0.40、1.00、2.00 mg/L 的系列标准工作溶液,现用现配。
1.3 样品前处理
低温下准确称取1.000 g(精确至0.001 g)已粉碎均匀的样品于样品管中,加入约40 mL 的超纯水,立即盖紧密封,超声提取3 次,每次提取10 min,每次提取间隔为1 h,以防止其内部液体温度过高产生膨胀,继而顶出密封盖,从而造成样品损失。于25~40 ℃放置48 h,使样品充分水解(应充分振摇,使样品无结块,振摇时应按住样品管盖,防止样品溢出),再把样品管放入冰箱于4 ℃冷藏2 h后,以10 000 r/min 冷冻离心10 min,取出上清液,在样品管中加入超纯水10 mL,振摇,冷冻离心,将取出的上清液合并后,加入10 mL 正己烷,充分振摇,静置分层后舍去正己烷,水层经过0.45 μm 的水系过滤膜过滤,滤液定容至100 mL,加入氢氧化钠0.2 g 使氢氧化钠质量浓度为2 g/L,冷藏,待混匀,待测。
1.4 加标样品制备
称取6 个均匀样品1.000 g(精确至0.001 g)于样品管中,分3 个批次加入50 mg/L 氰化物标准溶液0.2、0.7、1.0 mL,同1.3 方法进行处理,分别得到质量浓度为0.1、0.35、0.5 mg/L 的加标样品。
1.5 仪器工作条件
进样时间:80 s;冲洗时间:90 s;探测峰起始值:0.01 AbS;峰形宽度:30%(5%~100%);加热器温度:155、40 ℃;检测波长:600 nm。
2 结果与讨论
2.1 氢氧化钠加入时机的优化
氢氧化钠的加入时机,分为在提取初和提取后加入。在提取初加入氢氧化钠可以更好的保留样品中氰化物的含量,但实际操作中,在提取初加入氢氧化钠易致使样品“胶质化”严重,溶液不易经0.45 μm 的水系过滤膜过滤,故选择在低温提取后加入氢氧化钠调节pH。
2.2 萃取剂及其用量的选择
2.2.1 萃取剂
对于油脂含量较低的样品,可直接经0.45 μm的水系过滤膜过滤测定,但杏仁中油脂含量高达50.33%[14],易造成滤膜堵塞,不宜过滤。在了解杏仁中脂肪及蛋白质的含量及特性[15]后,选择易过滤样品,经正己烷处理后测定,结果数据见表1。由表1 可知,样品加标回收率为98.0%~104.7%,符合测定要求,正己烷对于测定结果并未产生偏差影响,因此选用正己烷为萃取剂。
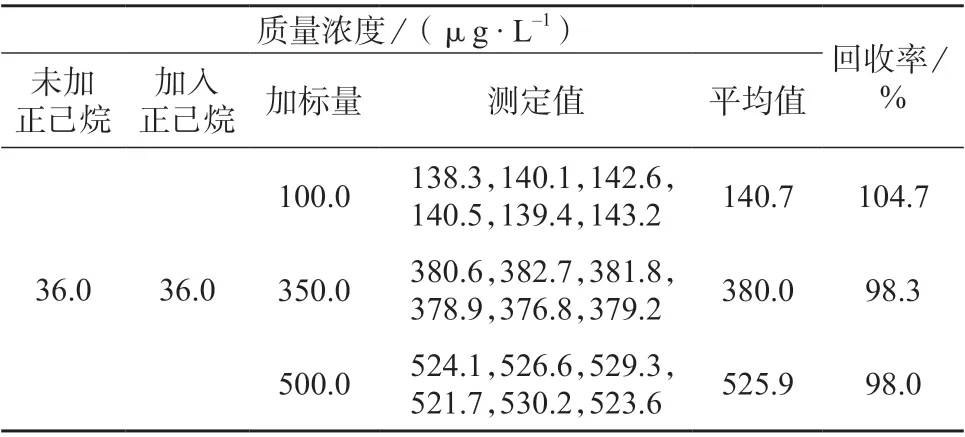
表1 加入正己烷后样品测定结果
2.2.2 正己烷用量
分别采用5、10、20 mL 正己烷完成萃取过程,所得试验结果见表2。由表2 可知,样品加标回收率为98.3%~97.3%,符合测定要求。经t检验,在此样本量下,分别加入5、10、20 mL 正己烷后,结果差异均无统计学意义(P>0.05),但在实际操作中,为保证油脂量高样品的萃取效率,推荐加入10 mL 正己烷进行萃取。
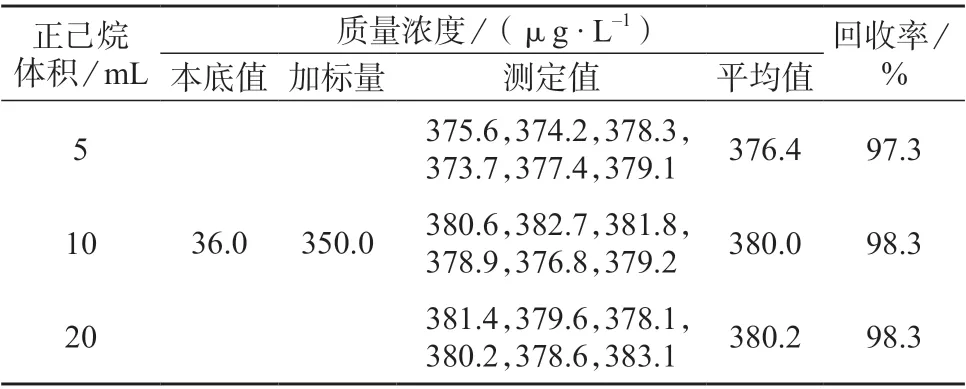
表2 加入不同体积正己烷后样品测定结果
2.3 样品放置时间及温度选择
参照食品风险监测中气相色谱法的前处理条件,为了使样品能够水解地更加充分,将样品于25~40 ℃条件下放置48 h 密封水解,样品充分水解后,放至冰箱冷藏2 h 再离心,可使样品更好地脂水分离,且提高了正己烷的萃取效果,因此样品水解温度为25~40 ℃,放置时间为48 h。
2.4 线性方程与检出限
2.4.1 线性方程
在1.5 仪器工作条件下测定氰化物系列标准工作溶液,以氰化物质量浓度(x)为横坐标,吸光度(y)为纵坐标绘制标准工作曲线,若取0~2.00 mg/L浓度区间,线性方程为y=0.000 64x+0.002 36,相关系数(r)为0.999 4;若取0~1.00 mg/L 浓度区间,线性方程为y=0.000 66x-0.000 55,相关系数(r)为0.999 9。比较发现,浓度过高易造成峰与峰之间的距离过小,峰面积叠加掩盖,导致曲线r值降低,因此连续流动分析法测定氰化物标准工作曲线的线性范围为0~1.0 mg/L,超出范围的样品需稀释后进行测定。
2.4.2 检出限
采用连续流动分析法测定空白样品11 次,若杏仁取样为1.000 g,定容100 mL 时,用2.764 倍标准差计算检出限,检出限为0.05 mg/kg,定量限为0.15 mg/kg。
2.5 回收率与精密度
在已知含量的样品溶液中加入1.4 中低、中、高浓度的加标样品溶液,在1.5 仪器工作条件下重复测定6 次,试验结果见表3。由表3 可知,平均回收率为98.0%~104.7%,测定结果的相对标准偏差为0.56%~1.34%,表明该方法具有较高的准确度和精密度,满足检测要求。
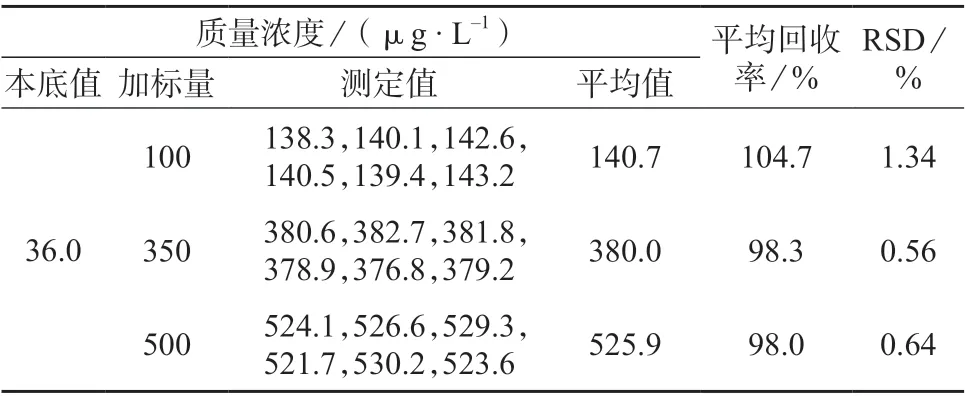
表3 加标回收率和精密度试验结果
2.6 比对试验
连续流动分析方法与气相色谱法从以下几个方面进行比对:
(1)开机稳定时间、样品用量及稳定性。连续流动分析仪开机到稳定耗时较短,从进样蒸馏到测定仅需约0.5 h,全程密闭蒸馏加试剂,连续进样后,每个样品分析时间约为2 min,每次进样为2 mL,在分析过程中序列可以连续加入后续样品。气相色谱仪稳定耗时较连续流动分析仪长,每个样品分析时间约为10 min,每次进样为10 mL,且在现阶段技术条件下,样品序列编辑运行后不能随时加入后续样品,需重新编辑序列及测定标准。样品量少时,二者耗时区别不大,随着样品量增加,连续流动法测定耗时明显较气相色谱法短。连续流动法在样品提取后加入了氢氧化钠以保留氰基,使样品中被测物质在水中稳定性增加,因此从耗时耗量及样品稳定性上看,连续流动法优于气相色谱法。
(2)线性范围。连续流动法测定过程中氰化物浓度过高易造成峰过大,峰间距过小,峰面积叠加掩盖,从而导致测定值准确度低,因此连续流动分析法测定氰化物的质量浓度应在0~1.0 mg/L 范围内,超出线性范围的样品需稀释后测定。气相色谱法的线性范围超过1.0 mg/L 后,相关系数降低,在0~1.0 mg/L 质量浓度范围内,连续流动法的r值为0.999 9,优于气相色谱法r值(0.998 6),因此在此浓度范围内连续流动法优于气相色谱法。
(3)检出限。气相色谱ECD 检测器对氰基响应灵敏度高,检出限较连续流动法低。虽气相色谱法检出限优于连续流动法,但两法检出限均能满足检测要求。
(4)回收率。两种方法的回收率均能满足检测要求,但气相色谱法回收率跨度远大于连续流动分析法,且方法的不可控因素更多(如氯胺T),因此,在0~1.0 mg/L 质量浓度范围内连续流动法回收率要优于气相色谱法。
(5)精密度。在0~1.0 mg/L 浓度范围内,对两种方法的检测结果进行用F检验,低、中、高浓度组的F值均大于F值表中F(5,5)=5.05,即连续流动分析法的精密度优于气相色谱法。
连续流动分析法与气相色谱法比对结果如表4所示。
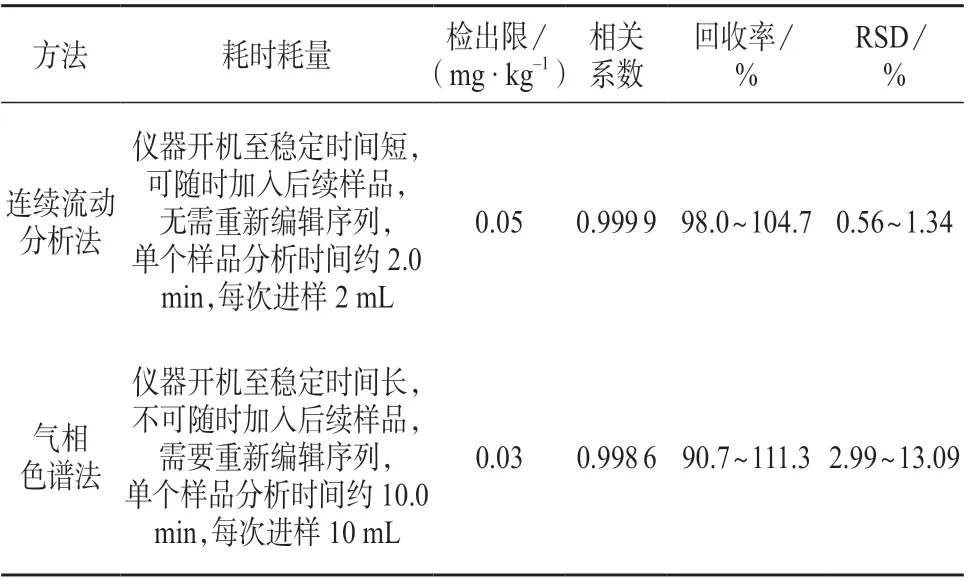
表4 比对试验结果
3 结语
建立了连续流动分析法测定杏仁中的氰化物。样品超声过程应密封且时间不能连续过长,以防止样品过热造成喷溅;样品水解后离心冷藏(4~8 ℃)时间应在30 min 以上,利于油脂分离;样品经正己烷萃取,并在滤液中加入氢氧化钠,以增加氰化物在水溶液中的稳定性;试剂在测定过程中均在较密闭空间内。连续流动分析的精密度、加标回收率等较气相色谱法的误差小,虽检出限较气相色谱法略高,但在实际工作中能满足检测要求,因此连续流动仪测定杏仁中氰化物的方法整体优于气相色谱法,其在线分析时间更短,稳定性及安全性更好,适于推广应用。
猜你喜欢
中南民族大学学报(自然科学版)(2022年3期)2022-05-08 03:51:04
节能与环保(2022年3期)2022-04-26 14:32:44
Journal of Literature and Art Studies(2021年11期)2021-03-03 18:18:09
天津化工(2019年6期)2019-12-10 00:40:08
创新作文(1-2年级)(2017年6期)2017-11-30 18:49:03
电镀与环保(2017年3期)2017-06-23 08:24:52
西藏科技(2016年9期)2016-09-26 12:21:42
少年科学(2015年9期)2015-10-14 04:14:24
故事作文·高年级(2015年1期)2015-09-07 08:17:04
中国有色冶金(2014年6期)2014-08-10 12:29:04
