
基于宏基因组学的酸性森林土壤氨氧化微生物群落特征研究*
2021-10-11 02:07:26唐修峰王欣欣宋玉翔刘林梦张焕朝王保战
土壤学报 2021年5期
唐修峰,秦 华,匡 璐,王欣欣,宋玉翔,高 豪,刘林梦,任 一,单 军,张焕朝,王保战,5†
(1. 南京林业大学林学院,南京 210037;2. 土壤与农业可持续发展国家重点实验室(中国科学院南京土壤研究所),南京 210008;3. 浙江农林大学亚热带森林培育国家重点实验室,杭州 311300;4. 上海美吉生物医药科技有限公司,上海 201318;5. 南京农业大学生命科学学院,南京 210095)
硝化作用被认为是全球氮循环的关键步骤,是由微生物驱动通过亚硝酸根,将氨氧化成硝酸根的过程[1]。而氮素作为森林生态系统中主要的限制性营养元素[2],其硝化作用产生的硝酸盐更易通过淋溶作用导致氮素流失和地下水污染等,此外硝酸根还可通过反硝化作用产生重要的温室气体氧化亚氮(N2O)。自1892年Winogradsky[3]发现氨氧化细菌(Ammonia-oxidizing bacteria,AOB)以来,人们一直认为硝化过程由两类微生物接力完成,分别为AOB催化的氨氧化过程和亚硝酸盐氧化细菌(Nitrite-oxidizing bacteria,NOB)催化的亚硝酸氧化过程,其中氨氧化过程被认为是硝化作用的限速步骤[4]。2005年,美国科学家David A. Stahl团队[5]从海洋中分离出第一株氨氧化古菌(Ammonia- oxidizing archaea,AOA),证明除细菌外,古菌也能够催化地球上的氨氧化过程。2015年,Daims[6]和van Kessel[7]等均发现了具有氨氧化活性的硝化螺旋细菌(Nitrospira),并证明其可将 4NH+氧化成NO3-,称为完全硝化微生物(Complete ammonia oxidizer,comammox)。Comammox的发现打破了上百年来人们认为硝化过程必须由两类微生物接力完成的传统观念,使人们再次认识到硝化微生物的物种多样性和生理代谢机制的复杂。
酸性pH和低氨分子浓度是森林土壤的两个重要特征。一般认为酸性pH选择了严格嗜酸group 1.1a-associated AOA类群[8-10],且DNA-SIP(稳定性同位素示踪)实验证明该类AOA主导了森林土壤氨氧化过程[11]。但也有一些研究发现group 1.1b AOA可存在于酸性土壤[1,12],且能主导某些酸性农田土壤氨氧化过程[13],以及group 1.1b AOA在酸性森林土壤中也广泛存在,且在转录组水平发挥了较高的活性[14-15]。尽管关于AOB适应酸性pH及其相对应的低NH3环境的机制目前多为推测[16-17],但不少森林土壤中确实存在β-proteobacterium(β-变形菌门)中Nitrosospira(亚硝化螺旋菌)属AOB,而Nitrosomonas(亚硝化单胞菌)AOB较少检测到[18-20]。最近日本科学家又从pH 3.0左右的酸性茶园土壤中分离到耐酸γ-proteobacterium(γ-变形菌门)AOB菌株[21],然而森林土壤中是否存在耐酸 γ-proteobacterium AOB仍不清楚。此外最近备受关注的comammox,其在森林土壤中生理生态的研究也刚刚起步,而已报道的comammoxamoA基因PCR引物易产生非特异性扩增,可能导致其基于实时荧光定量PCR(qPCR)的丰度被严重高估[22]。土壤总DNA宏基因组测序由于不受PCR引物偏好性等限制,因此在土壤氨氧化微生物amoA基因相对丰度和群落组成的研究中更具优势。
马尾松林(Pinus massoniana)是我国南方广泛分布的主要人工林之一,具有高经济价值、耐干旱贫瘠等特点[23],其土壤为典型的酸性森林土壤。本文以浙江省建德市人工马尾松林土壤为研究对象,综合利用qPCR、凝胶电泳半定量和土壤总DNA宏基因组测序等手段,研究酸性森林土壤中AOA、AOB和comammox三种氨氧化微生物的丰度和群落组成等,以期加深对酸性森林生态系统中氨氧化微生物潜在生理生态学特征的认识。
1 材料与方法
1.1 供试材料
土壤样品于2017年4月采自浙江省建德市人工马尾松林(119°28′E,29°48′N),该地区属亚热带海洋型季风气候,海拔约200 m,年均气温17.4 ℃,年均降水1 600 mm。土壤类型为凝灰岩发挥的红壤,选择3个林龄均为55年的马尾松林(间隔约2 000 m),分别标记为M1、M2和M3。五点法采集0~20 cm土壤,每个马尾松林采集3个样品,样品分成两份,一份保存于–20 ℃用于提取土壤总DNA,另一份4 ℃保存用于土壤理化性质测定。
1.2 土壤理化性质测定
土壤含水量采用烘干法。pH采用电位法测量,水土比为5∶1。有机碳含量通过浓硫酸-重铬酸钾法测定。全氮和碱解氮的含量分别采用半微量凯氏定氮法和碱解扩散法。有效磷测定采用0.05 mol·L–1HCl和0.025 mol·L–1(1/2H2SO4)浸提—钼锑抗比色法。速效钾测定采用乙酸铵浸提—火焰光度法。经测定三个森林土壤pH为3.88~4.16,含水量173.1~203.4 g·kg–1,有机碳14.01~16.62 g·kg–1,全 氮 0.76~1.02 g·kg–1,碱 解 氮 123.24~142.81 mg·kg–1,有效磷6.34~10.49 mg·kg–1,速效钾56.12~60.15 mg·kg–1。
1.3 土壤DNA提取
土壤微生物总DNA提取采用Fast DNA Spin kit for soil试剂盒(MP Biomedicals,美国)。根据试剂盒操作说明提取土壤微生物总DNA,并使用Nanodrop ND-1000 超微量分光光度计(NanoDrop Technologies,Wilmington,DE,美国)和1%的凝胶电泳检测DNA浓度和质量,将DNA于–20 ℃保藏。
1.4 实时荧光定量PCR和凝胶电泳半定量分析
利用美国伯乐公司CFX96实时荧光定量PCR仪(Bio-Rad Laboratories,Hercules,美国)对人工马尾松林土壤中AOA、AOB和comammox的amoA基因进行定量分析。反应体系为20 μL:10 μL SYBR Premix Ex Taq(TaKaRa,日本),上下游引物0.5 μmol·L–1,2 μL(约1~10 ng)模板DNA,补充6 μL双蒸水。qPCR引物和反应条件见表1[22,24-26]。本实验每个样品设置三个重复,每个重复包含三个测试平行。将含有AOA、AOB和comammoxamoA基因的单克隆质粒DNA梯度稀释成101~109的标准浓度作为qPCR标准曲线。通过琼脂糖凝胶电泳检查荧光定量PCR扩增产物的特异性。
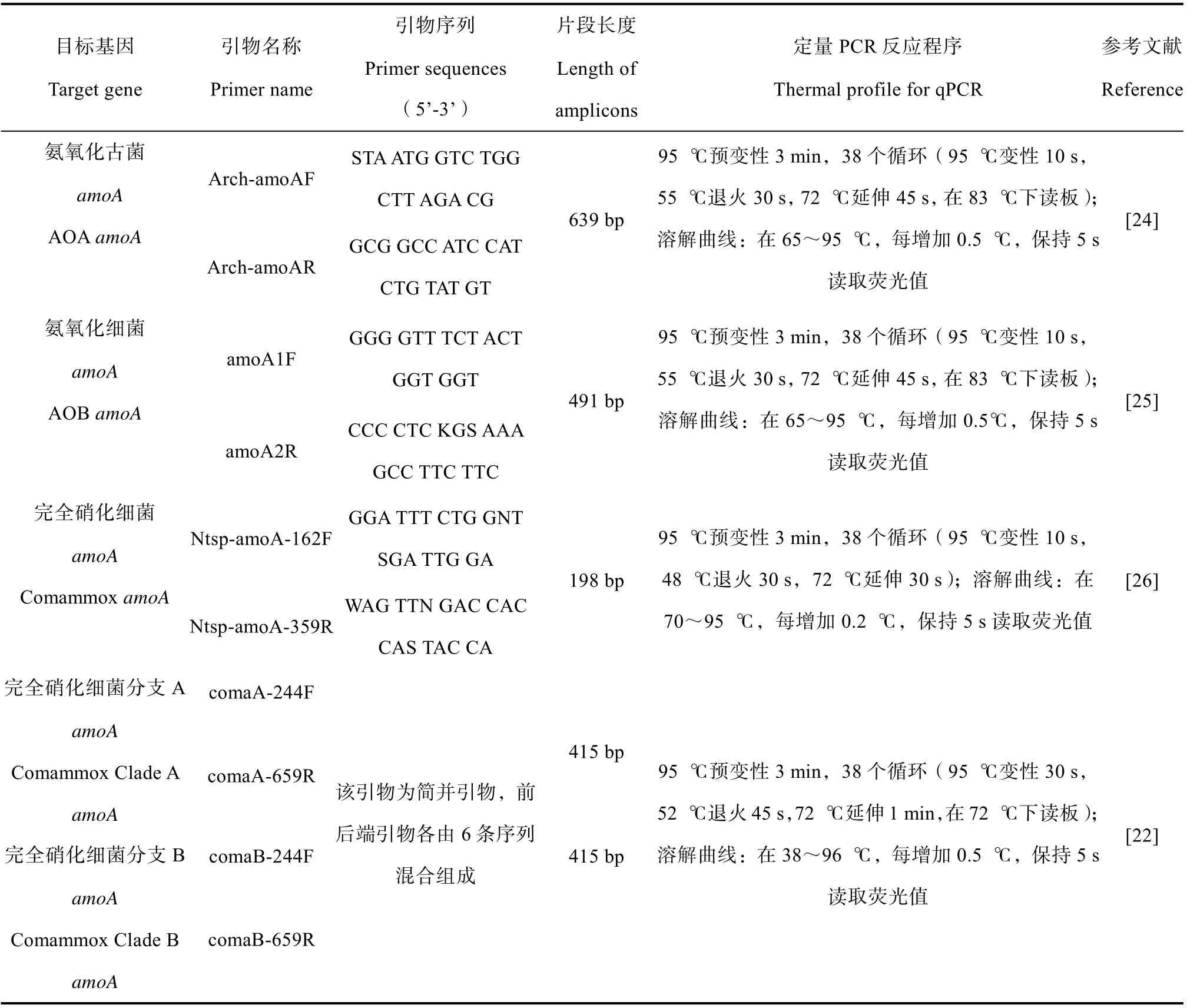
表 1 定量PCR扩增引物及反应条件 Table 1 Quantitative PCR amplification primers and their reaction conditions
本实验和已报道研究[27]均发现目前已知的comammox的amoA基因PCR引物易导致非特异性扩增,致使amoA基因丰度高估。因此利用凝胶电泳半定量法对qPCR结果进行数值初步矫正。即通过1.2%凝胶电泳和Bio-Rad公司的Quantity one v4.6.6软件检测待测样品qPCR产生的目标条带和非目标的相对灰度值,从而计算目标条带占泳道中总灰度值的百分比,进而用该百分比乘以qPCR检测结果,即得到样品中comammox的amoA基因实际拷贝数。同时我们将进一步用宏基因组中AOA、AOB和comammox三种amoA基因相对丰度,以及AOA和AOB的amoA基因的qPCR结果,进一步进行计算验证(见1.5)。
1.5 宏基因组测序和功能基因拼接及其相对丰度计算
委托上海美吉生物对土壤总DNA进行150 bp双端测序(Illumina HiSeq 2 000)。每个样品分别产生约48Gb的原始数据,然后通过SeqPrep(https://github.com /jstjohn/SeqPrep)去除末端测序接头(adaptor),然后利用Sickle(https://github.com/ najoshi/sickle)以默认参数对原始序列进行了质量控制和过滤,三个样品分别得到约42G bp高质量数据。使用metaSPAdes(version 3.10)宏基因组拼接软件对测序数据进行de novo从头组装,k-mer参数设置为21~53。使用Glimmer 3.0进行开放阅读框(ORFs)预测。以NCycDB数据库[28]中AOA和AOB的amoA基因以及Pjevac[22]、Daims[6]和Li[29]等文章中的comammox的amoA基因为参考序列,利用BLASTX与宏基因组拼接片段进行比对,提取Evalue ≤10–5且同源性高于70%的contigs作为amoA的候选序列,并进一步通过NCBI的nr数据库注释以及构建amoA进化树等手段进行验证去除错误注释的amoA基因序列。
分别采用两种方法计算宏基因组中AOA、AOB和comammox的amoA基因相对丰度。第一种方法,基于Mapped到amoA基因的HiSeq reads数统计。第二种方法,统计宏基因组中参与三种amoAcontigs拼接所用的HiSeq reads数,reads数高低反映了该类基因的相对丰度高低。即根据amoA基因参考序列,通过BLASTX与原始reads进行比对,统计其中Evalue ≤10–5,同源性≥70%,blast hit区域大于25个氨基酸长度的reads数目,通过比较三种amoA基因mapping的reads总数,确定三者间的相对丰度。
1.6 系统发育进化树构建
首先通过MOTHUR软件[30]按照95%序列同源性提取AOA、AOB和comammox的amoA基因的OTUs(operational taxonomic unit)代表性序列,然后利用MEGA 7.0软件的最大似然法(Maximum likelihood)构建三种氨氧化微生物amoA基因的进化树。
2 结 果
2.1 AOA、AOB和comammox amoA 基因丰度
qPCR结果显示酸性马尾松林土壤中AOA和AOB的amoA基因丰度分别为2.61×106copies·g–1和1.45×106copies·g–1(图1a)。基于引物Ntsp-amoA- 162F/359R和引物comaA/B-244F/659R的qPCR结果显示comammoxamoA基因拷贝数分别为4.14× 106copies·g–1和2.64×106copies·g–1。然而,凝胶电泳图显示两组引物的qPCR结果均有明显的非特异性扩增,导致comammox的amoA基因qPCR结果的高估(图1a)。通过凝胶电泳和Quantity one v4.6.6对comammox的amoA基因目标条带进行半定量估算后,得到两种引物扩增的comammoxamoA基因总拷贝数分别为 1.38×106copies·g–1和 1.47×106copies·g–1(图1a)。因此,氨氧化微生物功能基因amoA的相对丰度,comammox:AOA为0.53~0.56;comammox:AOB为0.95~1.01(图1a)。
基于宏基因组Mapped到amoA基因的HiSeq reads数目和参与amoAcontigs拼接的HiSeq reads数目分析发现,三类氨氧化微生物amoA基因相对丰度为:comammox:AOA约为0.55和0.29,comammox:AOB约为0.98和0.59(图1b)。
2.2 AOA、AOB和comammox的系统发育和群落组成
目前AOA主要包括Nitrosopumilus(group 1.1a)、Nitrosotalea(group 1.1a-associated)、Nitrososphaera(group 1.1b)和Nitrosocaldus(ThAOA)[31]。宏基因组分析发现,马尾松林AOA其主要类群为group 1.1b,约占AOA总丰度的85.66%~88.07%,其次为group 1.1a-associated,占11.93%~14.34%,未检测到group 1.1a和Nitrosocaldus类群(图2)。
AOB 主要分为 β-AOB(Nitrosospira和Nitrosomonas)和γ-AOB(Nitrosococcus,亚硝化球菌属)[32]。其中Nitrosospira包含cluster 1~4、cluster 0、Nitrosospirasp. Nsp65和Nitrosospirasp. Nsp57/58等分支[33-34]。Nitrosomonas包含N. europaea/N. mobilis、N. communis、N. marina、N. oligotropha、N. cryotolerans、Nitrosomonassp. Nm143和cluster 5等分支[35]。宏基因组分析显示,Nitrosospira是AOB的主要类群,占AOBamoA基因总丰度的51.71%~63.96%,主要隶属于cluster 3、0和Nitrosospira-like(图 3),而Nitrosomonas占 36.04%~48.29%,主要隶属于Nitrosomonas communis和Nitrosomonas oligotropha(图 3)。
目前报道的comammox均来源于Nitrospiralineage Ⅱ,且主要分为clade A和clade B两支[6-7],近期有研究将comammox clade A进一步分为两个单系群,clade A.1和clade A.2[36]。土壤宏基因组分析发现,酸性森林土壤中comammox的主要类群为clade B,占comammoxamoA基因总丰度的63.39%~71.39%,此外clade A.1占总序列数的28.61%~36.61%,未检测到clade A.2序列(图4)。
3 讨 论
马尾松林土壤属于典型酸性森林土壤。为了得到AOA、AOB和comammoxamoA基因准确丰度,本研究同时使用了qPCR、凝胶电泳半定量和宏基因组三种方法进行数据比较分析。qPCR和宏基因组均表明AOAamoA基因丰度约为AOBamoA基因的两 倍(图1)。而comammoxamoA基因的两套引物存在明显的非特异扩增,该结果与Beach和Noguera[27]研究结果一致(图1a)。通过凝胶电泳半定量对qPCR结果进行矫正以及宏基因组测序分析都发现,comammoxamoA基因拷贝数和AOB的amoA基因处于同一个水平,均为AOAamoA基因拷贝数的50%左右(图1)。然而,目前comammox的qPCR 定量仍处于起步阶段。而凝胶电泳半定量也存在一定缺陷,其反映是qPCR结束时目标电泳条带和非目标电泳条带的灰度值之比,无法真正反映qPCR反应过程中特异扩增和非特异扩增释放的荧光信号强度之比。此外,凝胶电泳半定量目标条带位置,仍可能包含非特异扩增的序列。而宏基因组技术目前成本较高,数据分析也较繁琐。因此,仍需进一步发展comammoxamoA基因qPCR更有效的引物,或选择comammox基因组中其他marker基因设计qPCR引物。
pH和NH3浓度是影响酸性土壤氨氧化微生物群落最重要的两个环境因子。自首株严格嗜酸AOA菌株分离以来[8],大量基于DNA-SIP实验证据表明group 1.1a-assocaited AOA主导了很多酸性土壤的氨氧化过程[9,11,37]。然而我们发现group 1.1b AOA也可以在某些酸性土壤中发挥着主导作用[13]。James I. Prosser团队[38-39]通过全球、区域和田块三个尺度研究发现group 1.1b AOA也存在于酸性土壤中,且亚枝cluster 11被认为嗜酸生长。本研究发现马尾松林土壤中group 1.1b AOA占据数量上的优势,而group 1.1a associated仅占12%(图2)。此外,酸性土壤中NH3而被大量离子化成NH4+,而目前认为氨氧化微生物的底物为氨分子而非铵离子[40],因此推测酸性土壤中group 1.1b AOA能够适应较低氨环境。
酸性土壤中AOB的贡献长期以来一直存在较大争议。在AOA发现前,一直认为AOB驱动了酸性土壤的氨氧化过程。但后来发现分离于酸性土壤的Nitrosospira和Nitrosomonas纯菌株都不能在实验室内的酸性pH条件下生长[16]。Prosser等[16-17]推测可能有三种原因:(1)土壤中存在微域中性或碱性pH环境;(2)AOB在土壤中形成生物膜或团聚体抵抗酸性pH;(3)很多AOB菌株具有尿素水解酶活性,能够通过利用尿素抵抗(酸性土壤中)低氨胁迫。我们认为第三种解释可能混淆了酸性pH和低氨浓度两种环境胁迫,通过利用尿素确实可以应对低氨寡营养胁迫,但该AOB菌株能够在酸性pH条件下生存,还必须同时能够抵抗低pH胁迫。推测James I. Prosser等当时实验选择的Nitrosospira纯菌株本身能够耐受酸性pH,它只需要在此基础上通过利用尿素应对低氨胁迫即可在酸性pH培养基中生长。这点可以通过Prosser等[16-17]两篇文章中推测出来,五株来源于酸性土壤的AOB纯菌株均可以利用尿素,但只有一株Nitrosospira菌株可以通过利用尿素在酸性pH培养基中生长。因此,利用尿素对以上来源于酸性土壤的五株AOB纯菌株来说是适应酸性pH的必要但非充分条件。2017年Hayatsu等[21]分离得到在不添加尿素条件下,能够在酸性pH中生长的AOB纯菌株γ-proteobacterium TAO100。然而目前β-proteobacterium AOB适应酸性土壤的分子生物学机制仍不清楚。。
作为最新发现的comammox,其在地球各个生境中的分布规律备受关注。目前发现comammox在土壤、沉积物、滩涂、植物叶表、地下水、饮用水以及废水处理系统均有分布[6-7,22,36]。本研究发现酸性森林土壤也存在丰度与AOB(每个AOB细胞平均含有1.5个拷贝的amoA基因)相当的comammox 类群(图1),其中clade B占comammox总类群的64%,而clade A约占36%。该结果和已报到森林土壤中comammox群落组成基本一致[22,36]。不同的是,Xia等[36]发现森林土壤中clade A主要隶属于clade A.2分支,而本研究发现马尾松林土壤中clade A主要隶属于clade A.1(图4)。以上数据暗示comammox无论在clade B还是clade A类群中都存在能够适应酸性土壤环境的物种。Michael Wagner课题组[41]继2015年发现第一株comammox纯菌株后,也明确了已知的comammox纯菌株对底物氨具有很高的亲和力,其亲和力高于所有已知AOB菌株和绝大部分土壤来源的AOA菌株,因此这可以从一定程度解释comammox如何适应酸性土壤中的低NH3胁迫。然而,comammox如何适应酸性pH胁迫仍不清楚。
4 结 论
本文利用qPCR、凝胶电泳半定量和宏基因组测序等技术研究酸性森林土壤氨氧化微生物的群落结构。qPCR显示AOA和AOB的amoA基因丰度为2.61×106copies·g–1和 1.45×106copies·g–1。而comammoxamoA基因的扩增引物产生明显的非特异性扩增,导致qPCR结果的高估。半定量和宏基因组分析发现,氨氧化微生物amoA基因相对丰度为:comammox:AOA=0.55,comammox:AOB=0.98,推算comammoxamoA基因真实丰度为(1.38~1.47)×106copies·g–1。此外,AOA主要类群为group 1.1b占其总丰度的88.07%,而经典嗜酸类群group 1.1a-associated仅占11.93%。AOB主要类群为Nitrosospira(63.96%),而Nitrosomonas为36.04%。comammox主要类群为clade B(63.39%),而clade A仅占36.61%且均隶属于clade A.1亚枝。
致 谢感谢浙江农林大学孙凯和张云晴等同学在样品采集和理化性质测试中给予的帮助!
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:28
福建基础教育研究(2019年8期)2019-05-28 08:39:51
中学生数理化·八年级物理人教版(2017年6期)2017-11-09 06:00:43
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
湖北农业科学(2015年12期)2015-08-13 08:51:53
安徽理工大学学报·自然科学版(2015年1期)2015-07-21 14:24:00
安徽农学通报(2015年8期)2015-07-02 01:37:32
亚热带资源与环境学报(2015年4期)2015-01-22 07:06:16
东北师大学报(自然科学版)(2014年1期)2014-02-27 08:02:15
亚热带资源与环境学报(2014年3期)2014-01-23 01:52:46
