
SGLT1/2双靶点抑制剂的研究进展
2021-10-09 08:15杨中兴贺岩杨龙陈建超孙铁民沈阳药科大学基于靶点的药物设计与研究教育部重点实验室制药工程学院沈阳006北部战区总医院药剂科沈阳0840
中南药学 2021年9期
杨中兴,贺岩,杨龙,陈建超*,孙铁民*(.沈阳药科大学基于靶点的药物设计与研究教育部重点实验室制药工程学院,沈阳 006;.北部战区总医院药剂科,沈阳 0840)
糖尿病是一种慢性代谢性疾病,特征为患者的血糖长期高于标准值。当胰腺产生不了足够的胰岛素或者人体无法有效地利用所产生的胰岛素时,就会出现糖尿病。糖尿病主要分为1 型糖尿病(type 1 diabetes mellitus,T1DM)和2 型糖尿病(type 2 diabetes mellitus,T2DM),T1DM 好发于青少年,主要是因为胰岛β细胞产生胰岛素不足导致的,有明显的临床症状,例如多饮、多尿、多食等[1]。T2DM 主要发生在中老年人中,比T1DM 更加常见,约占糖尿病的90%~95%,主要是由于胰岛素抵抗(胰岛素调控葡萄糖代谢能力)或胰岛素分泌不足导致,一般无明显的临床症状[2]。随着时间的延长,糖尿病会严重损害人体的心脏、血管、神经等健康系统。根据国际糖尿病协会预测,至2030年全球成人糖尿病数量将增加至5.52 亿,每年花费将近8250 亿美元,已成为全球威胁人类健康的三大慢性非传染性疾病之一[3-5]。
目前,已有众多药物可以用于糖尿病治疗,如胰岛素类似物、双胍类、磺酰脲类、格列奈类[6]。然而,传统降糖药物存在一些不良反应,如胰岛素类、磺酰脲类、格列奈类药物使用不当会引起低血压,双胍类药物易引起乳酸性酸中毒等[7]。这些不良反应均会影响糖尿病的治疗,因此迫切需要开发新型降糖药物来满足患者的需要。
钠-葡萄糖协同转运蛋白(sodium-glucose cotransporters,SGLTs)是一类在小肠和肾脏中表达的葡萄糖转运蛋白,其主要功能是介导葡萄糖的跨膜运输,已被美国食品和药品监督管理局批准为一类新型降糖药物,受到了各界广泛关注[8]。目前,人体已发现6 种SGLT 亚型,分别为SGLT1、SGLT2、SGLT3、SGLT4、SGLT5、SGLT6[9]。其中,对SGLT1 与SGLT2 两种亚型研究较深(见图1)。SGLT1 主要表达于小肠刷状缘和肾近曲小管的S2、S3 节段中,负责在肠腔中转运葡萄糖,重吸收肾近曲小管未被SGLT2 重吸收的10%葡萄糖[10]。SGLT2 主要分布在肾近曲小管S1 部位,完成约90%葡萄糖的重吸收[11]。SGLT1 和SGLT2的共同作用使尿液中的葡萄糖含量较少,若抑制SGLT1 和SGLT2 靶点,则可以减少小肠、肾脏对葡萄糖的重吸收,使过量的葡萄糖可以通过尿液排出体外,从而达到降低葡萄糖阈值效果。
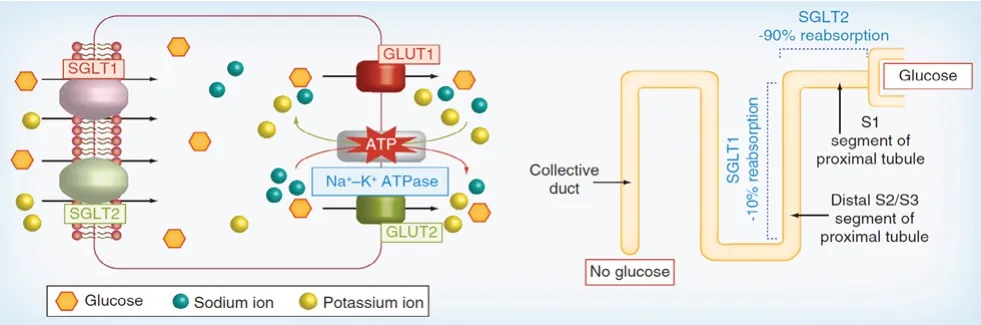
图1 SGLT1 和SGLT2 在肾单位中的位置和分布及其重吸收能力Fig 1 Location and distribution of SGLT1 and SGLT2 in the nephron and its reabsorption capacity
SGLTs 的研究发现,通过抑制肠道和/或肾脏葡萄糖重吸收来降低高血糖是一种有吸引力的治疗策略。若对SGLT1/2 多靶点进行双重抑制,既能够抑制SGLT1 而延缓肠道葡萄糖吸收,又可以抑制SGLT2 而增加葡萄糖的排出,从而有效地降低血糖水平。本文拟对该类新型抗糖尿病药物的研究进展进行综述。
1 SGLT1 与SGLT2 转运机制
葡萄糖在人体肾脏中的吸收和重吸收转运体主要分为两类,即葡萄糖转运蛋白(glucose transporters,GLUTs)和SGLTs。GLUTs 以易化扩散的方式转运葡萄糖,SGLTs 则以主动运输的方式转运葡萄糖[12]。
1.1 SGLT1 在肠道介导的葡萄糖吸收
SGLT1 在小肠刷状缘的上皮细胞膜中表达,主要功能为将肠道中的葡萄糖与半乳糖从内腔向上皮细胞转运,之后由GLUT2 介导的葡萄糖经基底外侧膜转运进入血液,从而进入体循环[13]。对SGLT1基因敲除小鼠的临床研究发现,餐后胃肠道会释放胰高血糖素样肽(GLP-1)和抑胃肽(GIP),刺激胰岛β细胞增加胰岛素的释放,进一步促进降糖作用[14]。SGLT1 抑制剂可以通过抑制肠道中的SGLT1 减少肠道中葡萄糖的重吸收,以及诱导胰岛素分泌刺激因子的释放,促进葡萄糖向远端位置的输送,从而维持葡萄糖的稳态。
1.2 SGLT1 与SGLT2 在肾脏介导的葡萄糖吸收
在肾脏中,葡萄糖则是由SGLT1 与SGLT2共同作用,其主要区别为SGLT2 为低亲和力、高转运的转运蛋白,在肾近曲小管的近端(S1、S2片段)中表达,可以对约90%的葡萄糖进行重吸收[15-16]。而SGLT1 则是高亲和力、低转运的转运蛋白,也可以在肾近曲小管的远端(S2、S3片段)中表达,吸收剩余10%的葡萄糖[17]。其机制主要为细胞外高浓度的Na+与SGLT1 负电荷位点结合,再与葡萄糖结合,形成Na+-载体-葡萄糖复合物,顺Na+的浓度梯度进入细胞后,在细胞内,SGLT1 与葡萄糖和Na+分离,使SGLT1 还原到原始构象状态,重新暴露其表面结合位点,以便再次与葡萄糖结合。Na+不断被细胞侧基底膜的Na+/K+-ATP 泵泵出,维持细胞内Na+浓度与胞外浓度差值,这种电位差产生的能量使葡萄糖逆浓度被转运到细胞内。细胞内的葡萄糖则由位于侧基底膜的载体GLUT2,经易化扩散转运进入到组织间隙液。SGLT2 则是结合一分子的葡萄糖,一分子的Na+,形成Na+-载体-葡萄糖复合物,进入到细胞中(见图2)。

图2 SGLT1 和SGLT2 转运葡萄糖机制Fig 2 SGLT1 and SGLT2 transport glucose mechanism
1.3 SGLT 抑制剂的开发
近年来已开发了多种SGLT 选择性抑制剂,从第一个SGLT2 选择性抑制剂——达格列净上市以来,随后多个SGLT2 选择性抑制剂相继上市(见表1)[18]。它们对SGLT2 选择性较高,对SGLT1 选择性较差。关于选择性抑制剂SGLT1,Mizagliflozin 是目前最具选择性的一种,对SGLT1的选择性是SGLT2 的300 倍[19]。并且Mizagliflozin被认为是不可吸收的肠道SGLT1 特异性抑制剂,显示出极低的生物利用度(F=0.02%),而在组织中没有任何明显的积累。由于Mizagliflozin 能增加腔内葡萄糖和水含量的能力,目前的临床试验正在研究使用其作为改善功能性便秘的候选药物。此外,新开发的化合物TP043883625,也是不可吸收的SGLT1 抑制剂,在啮齿动物的口服葡萄糖耐量试验中,显著降低了血糖波动[20]。

表1 SGLT2 上市药物及其SGLT1 抑制剂的研发Tab 1 SGLT2 listed drugs and development of SGLT1 inhibitors
SGLT1 选择性抑制剂通过对SGLT1 的抑制有助于降低心力衰竭的发生,对左心室肥厚和纤维化也起着重要作用[21]。但因具有过强的降血糖作用,不仅对肾脏远端、肾近端小管有抑制作用,也抑制了肠中葡萄糖的吸收和促进了尿葡萄糖的排泄,从而引起低血糖症等一些不良反应。
SGLT2 抑制剂可增强尿糖排泄来控制患者血糖水平,改善心力衰竭或慢性肾病的效果[22-23]。还有降低血压、调血脂、减重等疗效,并且不会发生低血糖的风险,但是SGLT2 抑制剂却有一些常见的不良反应,如泌尿生殖道感染、酮症酸中毒等[24-26]。因此研究人员正在探索减少SGLTs 抑制剂不良反应的新方法。
从代谢角度来看,在正常情况下,单独抑制SGLT1 的尿糖排泄效果较低。然而,当葡萄糖含量增加,或者肾小管前端SGLT2 被抑制,大量的葡萄糖则会向远端运输,SGLT1 对葡糖糖的重吸收比例则会增加到40%~50%,起到补偿作用的效果[27-28]。而对SGLT1/2 双靶点进行抑制,则可将更多的葡萄糖通过尿液排出体外,从而达到更好的降糖效果。故此,对SGLT1/2 双靶点抑制剂的研究开发具有较好的应用前景。
2 SGLT1/2 双重抑制剂研究进展
2.1 O-葡萄糖苷类
根皮苷(phlorizin,结构式见图3)是从苹果树的根皮中分离出来的第一个SGLTs 抑制剂,随后发现其是SGLT1 和SGLT2 的非选择性抑制剂[29]。研究表明服用1.0 g 以上的根皮苷会导致尿葡萄糖排泄(urinary glucose excretion,UGE),使空腹和进食的血浆葡萄糖水平趋于正常[30]。但因为根皮苷在肠道吸收差、生物利用度低以及被体内的β-葡萄糖苷酶降解为根皮素(phloretin,结构式见图3),阻碍了药物吸收到各组织中,故尚未进一步发展为抗糖尿病治疗药物。需要寻找发现更多的根皮苷衍生物,改善生物利用度差、容易被降解代谢失活等缺点。

图3 根皮苷、根皮素和三叶苷的化学结构Fig 3 Structure of phlorizin,phloretin and trilobatin
Wang 等[31]发现了根皮苷的异构体三叶苷(Trilobatin)也是一种O-葡萄糖苷类SGLT1/2 双靶点抑制剂,可明显地减少体内葡萄糖的吸收(结构式见图3)。为了研究三叶苷对SGLT1/2 的选择性,课题组将三叶苷以相同的方式与SGLT1 和SGLT2进行了分子对接(见图4)。结果发现,三叶苷与SGLT1 的相互作用和与SGLT2 的相互作用相似,结合位点处有许多相似的氨基酸残基,表明三叶苷对SGLT1 和SGLT2 没有选择性。此外国内外的一些研究表明,SGLTs 抑制剂可减少肿瘤细胞中葡萄糖的摄取,从而发挥抗肿瘤细胞增殖活性,根皮苷也显示出对大鼠肿瘤细胞显著的抗增殖特性[32-33]。根据根皮苷抗增殖特性,课题组对三叶苷的抗肿瘤作用进行了研究。实验观察到使用高浓度(超过50 μmol·L-1)的三叶苷促进了人肝母细胞瘤细胞(HepG2)的增殖,细胞的数量增加了35%,选择性地诱导了人肝母细胞瘤细胞的增殖,表明了三叶苷对HepG2 并没有抗增殖的特性。提示并非所有的SGLTs 抑制剂都能抑制肿瘤细胞的增殖,需要进一步研究,评估新型降糖药物的抗肿瘤潜力。

图4 三叶苷和根皮苷与SGLT1 和SGLT2 的相互作用Fig 4 Interaction of trilobatin and phlorizin with SGLT1 and SGLT2
Tsujihara 等[34]以根皮苷为先导化合物,进行结构改造,六元糖环和两个苯环之间的连接基团是必须的,把A 环酚羟基甲基化得到化合物4(见图5),对SGLTs 仍有一定活性。之后用苯并呋喃环代替了A 环,B 环引入甲基发现活性较好。为了克服消化道中β-葡萄糖苷酶对化合物的水解,将六元糖环上的羟甲基酯化,进一步改善药物在体内的活性并且增加对β-葡萄糖苷酶的稳定性,基于此设计想法,得到了SGLT1/2 双靶点抑制的前药衍生物(T-1095),其在体内可代谢为T-1095A[35](见图5)。
Holbrook,1982年,主张把消费体验分为功能型体验和享乐型体验(主要是研究对象不同),并在此基础上,主要以享乐型体验为蓝本,创造出重要的4Es理论。

图5 以根皮苷为先导化合物的结构改造Fig 5 Structural modification with phlorizin as the leading compound
Oku 等[36]对T-1095 进行了代谢方面的研究,T-1095 通过口服吸收进入循环系统,被代谢为活性形式T-1095A,抑制肾脏中SGLTs 的活性,增加糖尿病动物的尿葡萄糖排泄,从而降低血糖水平。研究发现T-1095A 的抑制活性(SGLT1:IC50=0.20 µmol·L-1,SGLT2:IC50=0.05µmol·L-1)与根皮苷的抑制活性(SGLT1:IC50=0.16 µmol·L-1,SGLT2:IC50=0.16µmol·L-1)相当,但T-1095A 的活性却是T-1095(SGLT1:IC50=22.8 µmol·L-1,SGLT2:IC50=2.3 µmol·L-1)的6~120 倍。尽管化合物T-1095A 活性较好,但是仍然没有克服O-葡萄糖苷类似物药代动力学不稳定的缺陷,无法成药。
Frick 等[37-38]在专利中报道了一类新的杂环氟糖苷衍生物SGLT1/2 抑制剂(7)(结构式见图6),并对此结构进行了改造。在研究中发现,在六元糖环的C5 位有氟原子的取代效果较优,并对中心的杂环选取了噻吩、呋喃、吡咯、吡唑等进行了研究,发现吡唑环对其活性较好。之后对取代基R1和R2改造,R1选取了羟基、三氟甲基、甲基、氢原子等,R2选取了甲氧基、卤素、乙基、甲基等,发现当R1为三氟甲基,R2为甲氧基时活性较好,最终合成了化合物SAR-7226,对人的SGLT1/2 的IC50分别为0.029、0.088 µmol·L-1[39]。

图6 杂环氟糖苷衍生物的结构改造Fig 6 Structural modification of heterocyclic fluoroglycoside derivatives
2.2 C-葡萄糖苷类
除了常见的氧-糖苷,碳-糖苷结构在生物活性方面也具有较多的优点,如比氧-糖苷具有更高的稳定性,在体内不易被水解酶降解等,因此碳-糖苷成为结构优化的有效策略。
Lexicon 制药公司以碳-糖苷为结构基础,研发出了化合物LP-925219(结构式见图7),此化合物是小鼠SGLT1/2 的有效抑制剂,其IC50值分别为22.6、0.5 nmol·L-1[40]。通过分别给野生型(WT)小鼠,SGLT1基因敲除、SGLT2基因敲除和SGLT1/2基因双敲除的小鼠饲喂LP-925219(10 mg·kg-1),并检测其24 h 的尿糖排泄。结果发现,SGLT1/2基因双敲除的小鼠UGE 是最大的,SGLT2基因敲除、SGLT1基因敲除和WT 小鼠分别是SGLT1/2基因双敲除小鼠UGE 的30%、2%和0.2%。每日两次以60 mg·kg-1剂量给药可以使SGLT1基因敲除、SGLT2基因敲除和WT 小鼠的UGE 升高至SGLT1/2基因双敲除小鼠的UGE水平。结果表明,口服LP-925219 抑制剂可以使哺乳动物的UGE 在24 h 内达到最大,与选择性SGLT2 抑制剂相比,化合物LP-925219 能更好地控制糖尿病患者的血糖。
Lexicon 制药公司随后以LP-925219 为基础,用乙氧基代替甲氧基,得到了活性较好的索格列净(sotagliflozin,见图7),对SGLT1 和SGLT2的IC50值分别为0.0018、0.036 μmol·L-1,于2019年4月26日获欧洲药物管理局(European Medicines Agency,EMA)批准上市,且为唯一上市的SGLT1/2 双靶点抑制剂的药物,在中国处于Ⅰ期临床试验状态[41]。

图7 LP-925219、sotagliflozin 的化学结构Fig 7 Structure of LP-925219 and sotagliflozin
Sands 等[42]评估了索格列净与胰岛素联合使用治疗T1DM 的有效性。选取33 例患者,其中17 名患者使用索格列净联合胰岛素治疗,16 名患者使用安慰剂联合胰岛素进行治疗。在索格列净治疗组中,注射胰岛素的剂量,与安慰剂组治疗相比剂量降低32.1%(P=0.007),同时测得平均每日血糖较低,为148.8 mg·dL-1,糖化血红蛋白(HbA1c)水平降低0.55%。而安慰剂组胰岛素的使用剂量比基线减少了6.4%,平均每日葡萄糖为170.3 mg·dL-1,HbA1c 水平下降0.06%。作为胰岛素的辅助药物,索格列净可改善血糖控制,减少胰岛素剂量,并且不会增加T1DM 患者血糖。
在肾功能不全的患者中,SGLT2 选择性抑制剂疗效不佳,故此Goodwin 等[44]以索格列净为先导化合物进行了结构改造,设计出了一种由于口服生物利用度差而停留在胃肠道内的SGLT1/2 抑制剂化合物LX2761,在胃肠道内局部抑制SGLT1而不影响肾脏中SGLT1/2(结构式见图8)。保持木糖结构,对双芳基苷元部分进行改造:① 在近端芳环上取代卤素原子X;② 改变远端芳环的连接原子Y 和链长度n;③ 改变链末端的基团Z。发现用甲基取代卤素X 基团,对SGLT1 活性更好。连接原子Y 用亚甲基取代氧原子,对SGLT1或SGLT2 抑制活性相似。链末端的基团Z 的改变对活性影响较大,官能团不同其口服吸收也不同。当以碱性胺为取代基团时,口服吸收减少,由此设计了一系列具有酰胺和胺类化合物,其中化合物LX2761 对SGLT1 和SGLT2 均有较好的活性,抑制活性IC50分别为2.2、2.7 nmol·L-1,并在胃肠道表现出特定的SGLT1 抑制作用。

图8 LX2761 的设计策略Fig 8 Design strategy of LX2761
Kakinuma 等[45]合成了化合物13,并对其进行了改造(结构式见图9)。当A 环上取代基R1被2-OH,R2被4-CH3取代时,其效能得到了增强。在B 环上讨论了不同的芳基取代基的影响,当B环为苯并环丁烷,A 环为2-羟基-4-甲基苯基时,得到了化合物14,显示出非常高的抑制活性,对SGLT1 和SGLT2 的IC50值分别为4 nmol·L-1与1 nmol·L-1。尽管化合物14 在SGLT1/2 上均表现出优异的体外效能,但酚类分子可能通过葡萄糖醛酸化产生Ⅱ期代谢问题[46]。研究发现当去除R1位的羟基,R2位用Cl 原子取代,得到的化合物15 也能保持较高的活性,对SGLT1 和SGLT2的抑制IC50分别为45、1 nmol·L-1[47]。化合物15 在小鼠、大鼠、狗和猴子中表现出较好的口服生物利用度,分别为99%、78%、107%、88%。在SD 大鼠中,化合物15 以剂量依赖性方式显著降低了血糖水平,在ZDF 大鼠中,化合物15 降血糖作用时长可长高达24 h。

图9 结构式13 的构造优化Fig 9 Structural optimization of structural 13
Jensen 制药在2014年发布的专利US90124-12B2 中发现了一类化合物16,作为SGLT1/2 抑制剂用于糖尿病的治疗,并对其进行了结构改造[48](见图10)。选取R1为OH、烷氧基、芳杂环或卤素,R2为氢原子、甲基,R3为甲基、烷氧基、卤素进行研究,发现当R1为羟基,R2为氢原子,R3为甲基时,活性较优,对SGLT1 和SGLT2的抑制IC50分别为117、16 nmol·L-1。后又对其A 环进行了优化,选取了苯并呋喃环、苯并噻唑环、2-乙基噻吩环、2-(4-氟苯基)噻吩环进行研究,发现A 环为2-乙基噻吩环时活性较好,得到化合物18,对人体肾脏和肠道中的SGLT1 与SGLT2 的抑制IC50分别为97、4 nmol·L-1。

图10 化合物18 的设计策略Fig 10 Design strategy of compound 18
针对C-葡萄糖苷类的降糖药,诺华公司发现了licogliflozin(见图11),现处于Ⅱ期临床试验阶段,对SGLT1 和SGLT2 的抑制IC50值分别为20.6、0.58 nmol·L-1[49]。针对化合物licogliflozin,He等[50]研究了其对T2DM 和肥胖患者的体重、代谢参数与肠降血糖素激素的影响。共纳入130名患者,随机接受licogliflozin 或安慰剂治疗。第一项肥胖患者研究共纳入88 位患者,用150 mg qd 的licogliflozin 治疗肥胖症患者,12 周后,较安慰剂组体重降低了5.7%(P<0.001),并改善了代谢参数,餐后葡萄糖的波动幅度降低了21%(P<0.001),胰岛素水平降低了80%(P<0.001),胰高血糖素升高了59%(P<0.001)。第二项研究纳入42 位T2DM 患者,用licogliflozin 15 mg qd 治疗连续14 d,与安慰剂组相比,T2DM 患者的24 h 平均血糖水平降低了26%(P<0.001),总GLP-1 提高了54%(P<0.001),PYY 提高了67%(P<0.05),而GIP 水平降低了53%(P<0.001)。licogliflozin治疗可明显减轻体重,各种代谢参数和肠降血糖激素发生了有利的变化。
Lee 等[51]报道了化合物HM41322(见图11),并与达格列净进行对比。HM41322 在体外对人的SGLT2(IC50=5.6 nmol·L-1)的选择性是SGLT1(IC50=54.6 nmol·L-1)的10 倍,其中对SGLT2抑制作用与达格列净(IC50=2.9 nmol·L-1)类似,但是却有着比达格列净更好的SGLT1 抑制性(IC50=920.4 nmol·L-1)。在小鼠的药代动力学研究中发现,在单次口服剂量为1 或3 mg·kg-1时,HM41322 的最大血浆浓度分别为439 和1830 ng·mL-1,1 mg·kg-1剂量时作用与达格列净(Cmax=457 ng·mL-1)类似,并且可以在30 min内快速吸收。化合物HM41322 还可通过抑制肾脏中的SGLT1 和SGLT2 来达到尿糖排泄最大化,在3 mg·kg-1的剂量下,24 h 内正常血糖小鼠的最大尿糖排泄为(19.32±1.16)mg·g-1,比达格列净(10.70±1.16)mg·g-1更加有效。
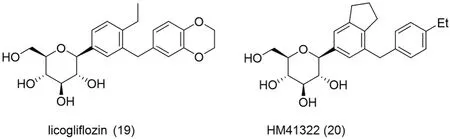
图11 licogliflozin、HM41322 的化学结构Fig 11 Structure of licogliflozin and HM41322
有报道指出C-F 键的长度(1.39 Å)与C-O键的长度(1.43Å)非常接近,氟可以充当羟基的等排体[40]。此外,早期的SAR 研究表明,C-芳基葡糖苷的C5 位可进行结构修饰[52]。因此,Xu等[53]对C-葡萄糖苷改造,用氟原子取代C5-羟基,以构建C5-氟己糖糖苷的SGLT 抑制剂,如化合物22(见图12,其中Ar 代表通用的苷元部分)。基于此设计合成了化合物23,其对SGLT1(IC50=43 nmol·L-1) 和SGLT2(IC50=9 nmol·L-1)有着较好的抑制效力。在SD 大鼠的口服葡萄糖耐量试验(oral glucose tolerance test,OGTT)中对血糖波动有着强烈的抑制作用,当以10 mg·kg-1的口服剂量在db/db 小鼠和高脂饮食喂养的ZDF 大鼠中表现出明显的降血糖作用。

图12 新型SGLT1/2 双靶点抑制剂的设计Fig 12 Design of a new dual inhibitor of SGLT1/2
此课题组又基于原先的策略,对糖环以及苷元部分进一步修饰,糖环C5 位处的氢都被氟原子取代,合成了化合物25[54]。化合物25 具有双重抑制活性(SGLT1:IC50=96 nmol·L-1,SGLT2:IC50=1.3 nmol·L-1),在口服1 mg·kg-1和10 mg·kg-1的SD 大鼠的OGTT 中,对葡萄糖显示出强烈的抑制作用,当口服剂量为10 mg·kg-1时,能较好地控制db/db 小鼠餐后的血糖。
之后此课题组对于单氟取代的C-芳基葡萄糖苷进行了改造优化[55](见图13)。当固定R2为羟基,R3为H 时,R4为甲基时,对R1进行了优化。发现当R1为羟甲基时,SGLT1 和SGLT2 的抑制率分别为IC50=0.014 μmol·L-1,IC50=0.004 μmol·L-1,效果较好。选取R1为其羟甲基,R3和R4不变,R2选取了甲基、甲氧基、卤素等基团进行改造,发现并没有羟基取代时效果好。故此,保持R2为羟基。之后用甲基取代了R3的H,其对SGLT1 和SGLT2 的抑制IC50分别为0.015、0.007 μmol·L-1,效果较优。最后对R4进行取代,发现当其为甲氧基时,效果最好,得到化合物27,对SGLT1 的IC50值为0.009 μmol·L-1,对SGLT2的IC50值为0.008 μmol·L-1。
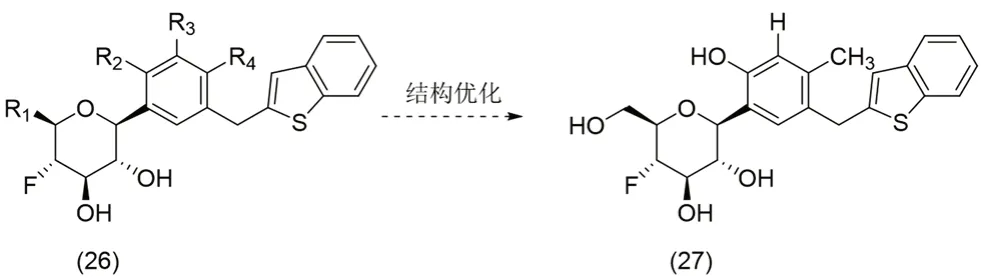
图13 单氟取代的C-芳基葡萄糖苷的优化Fig 13 Optimization of monofluoro substituted C-aryl glucoside
2.3 其他类
Park 等[56]发现了一种有效的新型合成小分子JP-2266(此结构尚未公开),是高度选择性的SGLT1/2 抑制剂,对SGLT1 和SGLT2 体外抑制IC50值分别为10.1 nmol·L-1和1.3 nmol·L-1,优于索格列净(36 nmol·L-1和1.8 nmol·L-1)。此外,给大鼠口服5 mg·kg-1的JP-2266 后,显示出与大剂量胰岛素治疗组(1.8 U·kg-1)相同的AUC0~4h的降低效果。当分别以10 mg·kg-1的JP-2266 和索格列净口服重复给药28 d 后,血糖分别降低49.4%和39.1%。另外,JP-2266治疗组的HbA1c 下降到6.4%,优于索格列净治疗组(7.2%),表现出较优的降血糖效果。
3 总结
控制血糖可以减缓糖尿病患者的病情,并降低其并发症。SGLTs 抑制剂通过抑制葡萄糖的重吸收,从而降低血液中葡萄糖的含量。但因其SGLT 选择性抑制剂有着不良反应的产生,限制了进一步的开发。SGLT1/2 双靶点抑制剂相比于选择性SGLT 抑制剂来说,有着较新的抑制机制,其降糖效果较好,能更好地控制餐后血糖水平,并可以减少不良反应的发生。
根皮苷为第一个SGLT1 和SGLT2 的非选择性抑制剂,但其生物利用度低、稳定性差,故此研究合成了一些根皮苷衍生物,以便改善其缺点。以根皮苷为其设计思路,合成了O-葡萄糖苷类衍生物,但仍然有药代动力学及稳定性差和对SGLT2 的选择性不足等缺点,之后用C-糖苷代替了O-葡萄糖苷,研究发现了一系列活性,稳定性较好的SGLT1 和SGLT2 双靶点抑制剂。不仅可以有效控制血糖,还有其他的临床发现,如减重,抗肿瘤细胞增殖等疗效。因此,新型降糖药物SGLT1/2 双靶点抑制剂有着较好的开发前景。
猜你喜欢
分子催化(2022年1期)2022-11-02
化学工业与工程(2022年1期)2022-03-29
中老年保健(2021年3期)2021-12-03
中国生殖健康(2020年7期)2020-12-10
第一财经(2019年8期)2019-08-26
中国塑料(2016年2期)2016-06-15
哈尔滨医药(2015年2期)2015-12-01
学习月刊(2015年14期)2015-07-09
医学研究杂志(2015年7期)2015-06-22
物理化学学报(2015年5期)2015-02-28
