
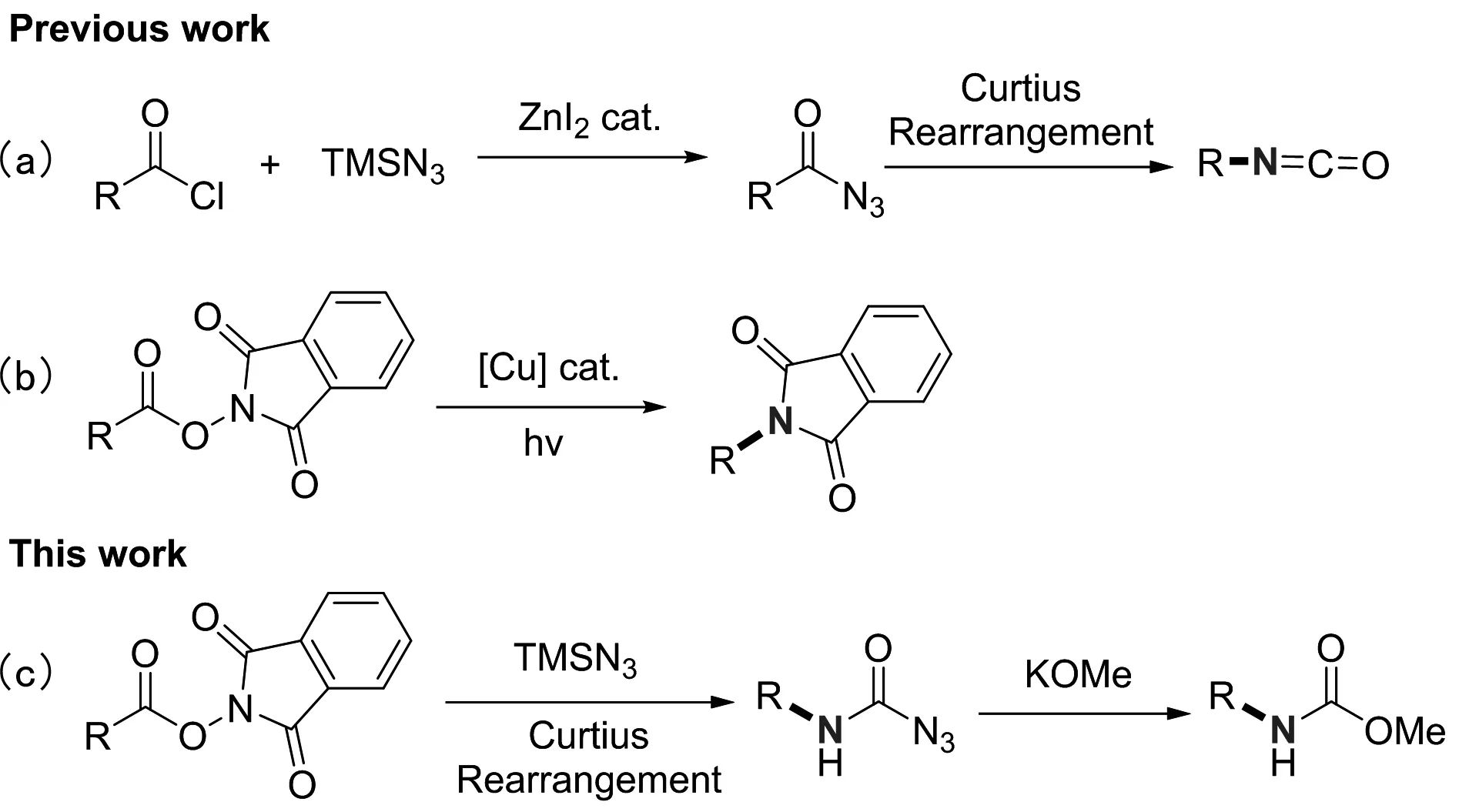
Synthesis of protected amines from N-hydroxyphthalimide esters via Curtius rearrangement
2021-10-08 01:11XuYingXuMengyu
中国科学技术大学学报 2021年2期
Xu Ying, Xu Mengyu
Department of Chemistry, University of Science and Technology of China, Hefei 230026, China
Abstract: An efficient and mild method to prepare carbamoyl azides from NHP (N-hydroxyphthalimide) esters and TMSN3 was developed. The structure of carbamoyl azide was confirmed by the X-ray analysis. Corresponding carbamoyl azides were converted into carbamates for isolation. This methodology allows an efficient access to primary, secondary, tertiary alkyl and aryl carbamates. Mechanistic studies reveal that Curtius rearrangement is responsible for the generation of carbamoyl azides.
Keywords: N-hydroxyphthalimide (NHP) esters; Curtius rearrangement; carbamoyl azides; amine derivatives; TMSN3
1 Introduction
C-N bond is widely occurring chemical bonds in chemical molecules, thus, the efficient construction of C-N bonds is of great importance in synthetic community. Textbooks reactions[1]such as Buchwald-Hartwig reaction, Ullmann coupling, Chan-Lam amination, Mitsunobu reaction, Curtius rearrangement, Ritter reaction and so on have been well-known for the construction of C-N bond. Among them, the rearrangement reaction of acyl azides reported by Curtius in 1894 comprises a classical method to construct C-N bonds from carboxylic acids[2]. In general, treating carboxylic acids or their derivatives with NaN3, TMSN3, DPPA or others is a useful method to prepare acyl azides[3-9]. However, before using TMSN3to yield acyl azide, it is necessary to transform carboxylic acids into activated acyl chlorides or anhydrides[9](Scheme 1(a)). The usage of activated carboxylic acid derivatives like acyl chlorides or anhydrides restricts the application of TMSN3in Curtius rearrangement.
It is well known that carboxylic acids and their derivatives can be easily converted into various functional groups to meet the needs of organic chemists. N-hydroxyphthalimide (NHP) esters, which are derived from carboxylic acids, have recently been intensively applied to organic synthesis as precursors to produce organic radicals[10-17]. In recent years, Fu group has developed decarboxylative C-N coupling to generate amine derivatives from NHP esters[17](Scheme 1(b)). In addition, high reactivity and stability of NHP esters grant them great value in nucleophilic substitution reaction. Taking advantage of NHP esters and TMSN3to prepare acyl azides has not been reported until now. Compared with acyl chlorides or anhydrides, NHP esters have obvious advantages in terms of easy operation on the bench and contain a wide range of functional groups owing to a mild preparation condition. Herein, we firstly reported in-situ generation of acyl azides from NHP esters and TMSN3, which led to the corresponding carbamoyl azides via Curtius rearrangement (Scheme 1(c)).
Scheme 1.ConstructionofC-Nbondfromcarboxylicacidderivatives.
2 Results and discussion
To evaluate the effects of reaction parameters, the control experiments were initiated wherein 4-phenylbutyric acid NHP ester (1a) and TMSN3were employed as reactants. Use of different solvents was found to be having a major impact on the yield. When THF or CH3CN was employed,2acould be obtained in 88% yield (Table 1, Entry 1-2). Using toluene as solvent led to significantly decreased yield (Table 1, Entry 3). The desired product was not observed when DMF was employed (Table 1, Entry 4). Lowering the reaction temperature only compromised the yield to a negligible extent (Table 1, Entry 5-6). What is more, exposure of the reaction in air only led to a slightly lower yield (Table 1, Entry 7). In order to investigate the effects of substituents on the NHP esters, other similar esters1a' and1a" were also employed (Table 1, Entry 8-9). It is obvious that the electron-withdrawing substituent on the NHP esters has a significant effect on the reactivity and a decreased yield of2awas observed. Besides, use of relatively electron-rich activated ester1a" did not improve the yield. Hence, we reckoned that N-hydroxyphthalimide esters have appropriate electronic properties for this reaction. It is worth noting that the additive is not required to promote the reaction.

Table 1. Effects of reaction parameters.
Having established the optimal reaction conditions, we next surveyed the generality of this reaction. For convenient isolation of the products, the resulted carbamoyl azides were converted to the corresponding carbamates by being treated with KOMe in MeOH.3awas successfully obtained in 85% yield by this strategy. Generation of3binstead of ring-opening product demonstrates that this reaction probably does not involve the radical intermediate. Iodine-bearing aryl group and protected amine are compatible in standard reaction conditions (3cand3e). Drug derivatives derived from Oxaprozin, Flurbiprofen and Gemfibrozil were readily converted to amine derivatives (3d,3fand3k). To further confirm the structure of the intermediate product2, the X-ray structure of2iwas established. All of these explored substrates have proved that not only primary alkyl carboxylic acid but also secondary and tertiary carboxylic acid derived NHP esters could react smoothly with TMSN3. It is noted that, aryl carboxylic acid derived NHP ester1lcould also give the corresponding product2lwith the yield of 53%, and2lwas conveniently converted to3lwith the treatment of KOMe in MeOH.
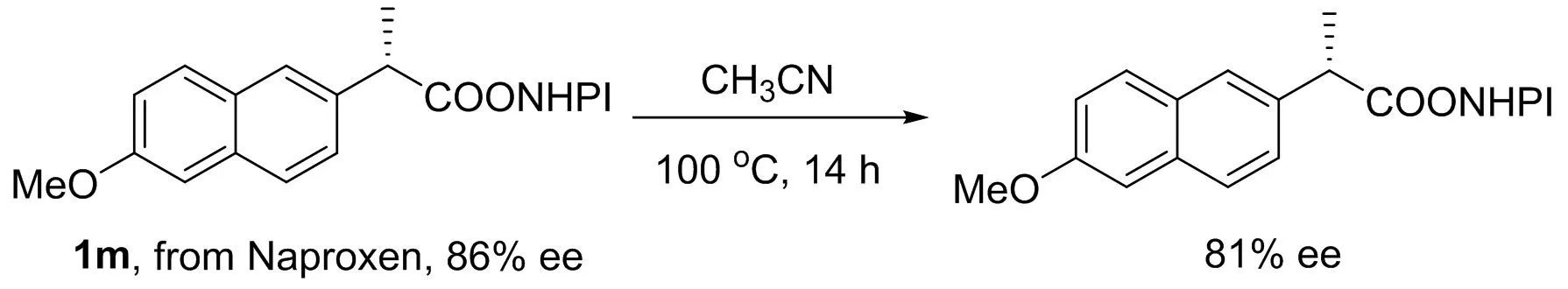
Based on the result of3b, we assumed that this reaction probably does not involve the radical intermediate. In order to further exclude the radical mechanism, the radical inhibitor TEMPO was employed. The reaction was not found to be suppressed at all and3acould be obtained with the yield of 85%. Byproduct4was detected by GC-MS analysis, which was in line with the result when TEMPO was not added (Scheme 2(a)). When NHP ester1m(86% ee) derived from Naproxen was applied under standard conditions, 41% yield of3m(0% ee) was acquired (Scheme 2(b)). We speculated that Curtius rearrangement of acyl azide should be responsible for the racemization(1)Without TMSN3, NHP ester derived from Naproxen only occurred racemization slightly.The ee of 1m was determined by HPLC. Column: AD-H; Flow rate: 1.0 mL/min; Mobile phase: Hexane/Isopropanol=90 / 10(V/V); Retention time: 33.553 min & 37.184 min.. Mechanistic studies showed that this reaction probably proceeded in the following manner: NHP ester reacted with TMSN3to generate4and acyl azide. Then, acyl azide underwent Curtius rearrangement to afford isocyanate, which further reacted with another TMSN3to produce the final carbamoyl azide (Scheme 2(c)). Deprotection of carbamoyl azide was also performed by treating2k, which was derived from Gemfibrozil, with lithium hydroxide aqueous solution. As expected, the free amine5was facilely accessed in 76% yield (Scheme 2(d)), suggesting an expedient approach to amines from carboxylic acids.
3 Conclusions
In conclusion,we have developed a method for the conversion of NHP ester to protected amines, which expands the application of TMSN3in Curtius rearrangement. The reaction enables the primary, secondary, tertiary and aryl carboxylic acid derivatives to be converted to amine derivatives. The mechanistic studies indicated that the reaction does not involve the radical mechanism, which is common for NHP ester in recent reports. This reaction has added a new content to Curtius rearrangement.

Table 2. Scope of NHP esters.
4 Experimental section
4.1 General procedure
An oven-dried screw-cap tube equipped with the stir bar was charged with 0.2 mmol corresponding NHP ester. The tube was vacuumed and backfilled with argon for three cycles. 0.6 mmol TMSN3and 1.0 mL CH3CN were added through syringes, and the tube was sealed with a teflon stopper and stirred at 100oC for 14 h. The reaction solution was concentrated under the reduced pressure, and 0.4 mmol KOMe and 1.0 mL methyl achohol was added. The reaction was then stirred at 75oC for 1 h. The reaction was quenched with NH4Cl (sat, aq), extracted with ethyl acetate. The organic layer was separated, dried over Na2SO4, concentrated under the reduced pressure to give a crude product, which was purified by silica gel column chromatography.
4.2 Characterization data for products
Methyl (3-phenylpropyl)carbamate (3a): obtained following the general procedure, 85% yield.1H NMR (500 MHz, Chloroform-d) δ 7.31-7.25 (m, 2H), 7.22-7.15 (m, 3H), 4.75 (s, 1H), 3.66 (s, 3H), 3.23-3.19 (m, 2H), 2.64 (t,J=7.7 Hz, 2H), 1.86-1.80 (m, 2H).13C NMR (126 MHz, Chloroform-d) δ 157.2, 141.5, 128.5, 128.5, 126.1, 52.1, 40.7, 33.1, 31.7. HRMS (ESI) Calcd for [C11H16NO2]+[M+H]+: 194.1181; Found: 194.1182.
Methyl ((2-phenylcyclopropyl)methyl)carbamate (3b): obtained following the general procedure, 80% yield.1H NMR (500 MHz, Chloroform-d) δ 7.28-7.22 (m, 2H), 7.18-7.12 (m, 1H), 7.06-7.01 (m, 2H), 4.89 (s, 1H), 3.66 (s, 3H), 3.33-3.13 (m, 2H), 1.83-1.73 (m, 1H), 1.34-1.29 (m, 1H), 0.98-0.85 (m, 2H).13C NMR (126 MHz, Chloroform-d) δ 157.2, 142.5, 128.5, 125.9, 125.8, 52.2, 45.4, 23.1, 22.1, 14.6. HRMS (ESI) Calcd for [C12H16NO2]+[M+H]+: 206.1181; Found: 206.1181.
Methyl (3-(2-iodophenoxy)propyl)carbamate (3c): obtained following the general procedure, 65% yield.1H NMR (500 MHz, Chloroform-d) δ 7.76 (d,J=7.6 Hz, 1H), 7.35-7.23 (m, 1H), 6.80 (d,J=8.1 Hz, 1H), 6.72 (t,J=7.5 Hz, 1H), 5.37 (s, 1H), 4.08 (t,J=5.8 Hz, 2H), 3.66 (s, 3H), 3.49-3.45 (m, 2H), 2.09-2.04 (m, 2H).13C NMR (126 MHz, Chloroform-d) δ 157.3, 157.2, 139.5, 129.6, 122.8, 112.0, 86.6, 67.5, 52.1, 39.0, 29.2. HRMS (ESI) Calcd for [C11H15INO3]+[M+H]+: 336.0097; Found: 336.0099.
Methyl (2-(4,5-diphenyloxazol-2-yl)ethyl)carbamate (3d): obtained following the general procedure, 51% yield.1H NMR (500 MHz, Chloroform-d) δ 7.64-7.55 (m, 4H), 7.43-7.30 (m, 6H), 5.54 (s, 1H), 3.83-3.53 (m, 5H), 3.05 (t,J=6.2 Hz, 2H).13C NMR (126 MHz, Chloroform-d) δ 161.5, 157.1, 145.7, 135.1, 132.4, 128.9, 128.8, 128.7, 128.7, 128.3, 128.0, 126.6, 52.2, 38.1, 28.8. HRMS (ESI) Calcd for [C19H18N2O3Na]+[M+Na]+: 345.1215; Found: 345.1212.
Benzyl 4-((methoxycarbonyl)amino)piperidine-1-carboxylate (3e): obtained following the general procedure, 81% yield.1H NMR (500 MHz, Chloroform-d) δ 7.39-7.30 (m, 5H), 5.11 (s, 2H), 4.89 (s, 1H), 4.25-3.99 (m, 2H), 3.68-3.65 (m, 4H), 3.02 - 2.92 (m, 2H), 2.00-1.83 (m, 2H), 1.36-1.29 (m, 2H).13C NMR (126 MHz, Chloroform-d) δ 156.3, 155.2, 136.7, 128.5, 128.1, 127.9, 67.2, 52.1, 48.2, 42.8, 32.3. HRMS (ESI) Calcd for [C15H21N2O4]+[M+H]+: 293.1501; Found: 293.1499.
Methyl (1-(2-fluoro-[1,1'-biphenyl]-4-yl)ethyl)carbamate (3f): obtained following the general procedure, 75% yield.1H NMR (500 MHz, Chloroform-d) δ 7.56-7.50 (m, 2H), 7.46-7.31 (m, 4H), 7.16 (d,J=7.9 Hz, 1H), 7.11 (d,J=11.6 Hz, 1H), 5.09 (s, 1H), 4.87 (s, 1H), 3.67 (s, 3H), 1.49 (d,J=7.0 Hz, 3H).13C NMR (126 MHz, Chloroform-d) δ 159.9 (d,J=248.2 Hz), 156.4, 145.5, 135.6, 131.0 (d,J=3.8 Hz), 129.1 (d,J=3.0 Hz), 128. 6, 128.0 (d,J=13.5 Hz), 127.8, 122.0, 113.7 (d,J=23.5 Hz), 52.3, 50.2, 22.5. HRMS (ESI) Calcd for [C16H17FNO2]+[M+H]+: 274.1243; Found: 274.1245.
Methyl (1,2,3,4-tetrahydronaphthalen-1-yl)carbamate (3g): obtained following the general procedure, 67% yield.1H NMR (500 MHz, Chloroform-d) δ 7.36-7.29 (m, 1H), 7.18-7.14 (m, 2H), 7.10-7.05 (m, 1H), 5.06-4.61 (m, 2H), 3.69 (s, 3H), 2.83-2.71 (m, 2H), 2.11-1.71 (m, 4H).13C NMR (126 MHz, Chloroform-d) δ 156.7, 137.6, 136.8, 129.2, 128.8, 127.4, 126.3, 52.2, 49.3, 30.6, 29.3, 19.9. HRMS (ESI) Calcd for [C12H15NaNO2]+[M+Na]+: 228.1000; Found: 228.1000.
Methyl (1-phenylpropan-2-yl)carbamate (3h): obtained following the general procedure, 57% yield.1H NMR (400 MHz, Chloroform-d) δ 7.30-7.28 (m, 2H), 7.25-7.14 (m, 3H), 4.56 (s, 1H), 3.97 (s, 1H), 3.64 (s, 3H), 2.84 (dd,J=13.5, 5.6 Hz, 1H), 2.69 (dd,J=13.4, 7.2 Hz, 1H), 1.11 (d,J=6.6 Hz, 3H).13C NMR (126 MHz, Chloroform-d) δ 156.4, 138.1, 129.6, 128.5, 126.6, 52.05, 48.0, 43.0, 20.4. HRMS (ESI) Calcd for [C11H15NaNO2]+[M+Na]+: 216.1000; Found: 216.1002.
Methyl (1-(3,5-dimethoxyphenyl)cyclopropyl)carbamate (3i): obtained following the general procedure, 36% yield.1H NMR (500 MHz, Chloroform-d) δ 6.57-6.23 (m, 3H), 5.43 (s, 1H), 3.77 (s, 6H), 3.66 (s, 3H), 1.40-1.12 (m, 4H).13C NMR (126 MHz, Chloroform-d) δ 161.0, 156.7, 145.6, 104.0, 98.1, 55.4, 52.2, 35.7, 18.2. HRMS (ESI) Calcd for [C13H17NaNO4]+[M+Na]+: 274.1055; Found: 274.1057.
Methyl (1-phenylcyclopentyl)carbamate (3j): obtained following the general procedure, 73% yield.1H NMR (500 MHz, Chloroform-d) δ 7.42-7.40 (m, 2H), 7.32-7.29 (m, 2H), 7.22-7.19 (m, 1H), 5.09 (s, 1H), 3.56 (s, 3H), 2.32-2.31 (m, 2H), 2.06-2.00 (m, 2H), 1.91-1.65 (m, 4H).13C NMR (126 MHz, Chloroform-d) δ 155.4, 145.3, 128.2, 126.7, 125.9, 66.1, 51.7, 39.0, 23.1. HRMS (ESI) Calcd for [C13H18NO2]+[M+H]+: 220.1338; Found: 220.1335.
Methyl (5-(2,5-dimethylphenoxy)-2-methylpentan-2-yl)carbamate (3k): obtained following the general procedure, 70% yield.1H NMR (500 MHz, Chloroform-d) δ 7.00 (d,J=7.4 Hz, 1H), 6.66 (d,J=7.5 Hz, 1H), 6.61 (d,J=1.6 Hz, 1H), 4.62 (s, 1H), 3.94 (t,J=6.0 Hz, 2H), 3.61 (s, 3H), 2.31 (s, 3H), 2.18 (s, 3H), 1.85-1.75 (m, 4H), 1.32 (s, 6H).13C NMR (126 MHz, Chloroform-d) δ 157.0, 136.6, 130.4, 123.7, 120.8, 112.1, 68.0, 52.7, 51.6, 36.9, 27.3, 24.5, 21.6, 15.9. HRMS (ESI) Calcd for [C16H25NaNO3]+[M+Na]+: 302.1732; Found: 302.1732.
Methyl (4-(tert-butyl)phenyl)carbamate (3l): obtained following the general procedure, two-steps, 48% yield.1H NMR (500 MHz, Chloroform-d) δ 7.33-7.28 (m, 4H), 6.66 (s, 1H), 3.76 (s, 3H), 1.30 (s, 9H).13C NMR (126 MHz, Chloroform-d) δ 154.3, 146.6, 135.3, 126.0, 118.7, 52.4, 34.4, 31.5. HRMS (ESI) Calcd for [C12H17NaNO2]+[M+Na]+: 230.1157; Found: 230.1157.
Methyl (1-(6-methoxynaphthalen-2-yl)ethyl)carbamate (3m): obtained following the general procedure, 41% yield.1H NMR (500 MHz, Chloroform-d) δ 7.75-7.61 (m, 3H), 7.38 (d,J=8.5 Hz, 1H), 7.17-7.06 (m, 2H), 5.08-4.97 (m, 2H), 3.90 (s, 3H), 3.66 (s, 3H), 1.54 (d,J=6.8 Hz, 3H). HRMS (ESI) Calcd for [C15H17NaNO3]+[M+Na]+: 282.1106; Found: 282.1107. The ee of 3m was determined by HPLC. Column: IC; Flow rate: 1.0 mL/min; Mobile phase: Hexane / Isopropanol=80 / 20(V / V); Retention time: 11.603 min & 14.800 min.
5-(2,5-dimethylphenoxy)-2-methylpentan-2-amine (5)obtained in 76% yield.1H NMR (400 MHz, Chloroform-d) δ 6.99 (d,J=7.4 Hz, 1H), 6.65 (d,J=7.5 Hz, 1H), 6.60 (s, 1H), 4.10 (brs, 1H), 3.92 (t,J=6.0 Hz, 2H), 2.29 (s, 3H), 2.17 (s, 3H), 1.84-1.74 (m, 4H). 1.29 (s, 6H).13C NMR (126 MHz, Chloroform-d) δ 157.0, 136.6, 130.4, 123.5, 120.8, 112.1, 68.1, 52.5, 37.2, 27.9, 24.6, 21.5, 16.0. HRMS (ESI) Calcd for [C14H24NO]+[M+H]+: 222.1858; Found: 222.1852.
Acknowledgments
This work was supported by the Fundamental Research Funds for the Central Universities (WK2060000014).
Conflictofinterest
The authors declare no conflict of interest.
Authorinformation
XuYingis currently a Master student in the Department of Chemistry under the supervision of Prof. Xiao Bin at University of Science and Technology of China. Her current research mainly focuses on organogermanium chemistry.
XuMengyu(corresponding author) received her PhD degree in Organic Chemistry from University of Science and Technology of China (USTC). She is currently a postdoctoral researcher at USTC. Her research interests include germanium-containing chemistry in organic synthesis and nucleophilic substitution of alcohols and carboxylic acids.

- 中国科学技术大学学报的其它文章
- Three-dimensional array materials for electrocatalytic water splitting
- LncRNA GIMA promotes hepatocarcinoma cell survival via inhibiting ATF4 under metabolic stress
- Effects of alanine-serine-cysteine transporter 2 on proliferation and invasion of hepatocellular carcinoma
- Histone methyltransferase SDG8 in dehydration stress
- The asymptotic properties of least square estimators in the linear errors-in-variables regression model with φ-mixing errors
- Electrodialysis to concentrate high-salinity solutions: The matching relation between cation- and anion-exchange membranes