
高温聚合物电解质膜燃料电池膜电极中磷酸分布及调控策略研究进展
2021-09-28 04:50张巨佳张劲王海宁相艳卢善富
物理化学学报 2021年9期
张巨佳,张劲,王海宁,相艳,卢善富
北京航空航天大学空间与环境学院,仿生能源材料与器件北京市重点实验室,北京 100191
1 引言
聚合物电解质膜燃料电池(PEMFC)具有清洁高效、高能量密度和可携带等特性,而被认为是最具潜力的能量转化设备1,2。由于电解质膜本身的特性,通常PEMFC的操作温度在80 °C以下(简称为低温聚合物电解质膜燃料电池,LT-PEMFC),使得其对CO等杂质气体的耐受性较差,水/热管理困难,热利用率不高,因此提高PEMFC的运行温度是解决上述问题的重要途径之一3-5。高温聚合物电解质膜燃料电池(HT-PEMFC)运行温度在140-200 °C之间,高的运行温度不仅提升了催化剂的反应动力学活性,同时降低了CO、SO2等杂质分子在铂(Pt)等贵金属催化剂表面的吸附,使电极对杂质分子的耐受能力大幅提升6-8。如图1a所示,当电池的操作温度≥ 200 °C时,即使阳极侧H2燃料中CO体积分数达到5%,但电池在0.6 V处的输出功率也不会明显下降(功率密度衰减≤ 15%)。同样,升高操作温度时阴极侧抗毒化也得到大幅提升8。如图1b所示,阴极侧通入0.01‰ SO2,HT-PEMFC在160 °C电流仅降低8%左右,而LT-PEMFC在80 °C电流却降低了82%9。因此,HT-PEMFC可以直接采用甲醇、甲酸等燃料重整气作为燃料9-12,大幅降低了聚合物电解质膜燃料电池对高纯氢气的高度依赖;并且对于阴极侧空气质量的要求相对较低9。同时,与燃料重整反应器相匹配的运行温度便于电堆和反应器的一体化设计,电池产生的余热也可以直接回收或提供给重整器使用,提高了整体系统的效率13-16。此外,水在HT-PEMFC运行过程中以气态水的形式存在有利于水的排出,而且不依赖水的质子传递机理使得反应气体无需加湿处理,从而简化了LT-PEMFC复杂的水/热管理系统17-19。因此,HT-PEMFC具有燃料选择广泛、系统设计简单、系统利用效率高等优势,是PEMFC未来重要的发展方向之一20-23。
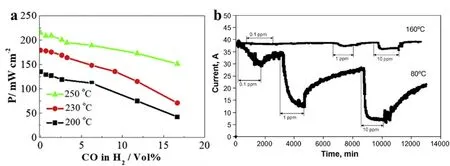
图1 (a) HT-PEMFC在0.6 V电压下的功率密度随温度和CO浓度的变化;(b) HT-PEMFC 160 °C和PEMFC 80 °C阴极侧SO2耐受性对比Fig. 1 (a) Power density measured at a cell voltage of 0.6 V, as a function of CO content in H2 fuel at different temperatures; (b) SO2 tolerance at cathode side of HT-PEM fuel cell at 160 °C, compared to LT-PEM fuel cell at 80 °C.
高温聚合物电解质膜(HT-PEM)作为HTPEMFC的关键材料,直接影响着燃料电池的输出性能和使用寿命。目前,HT-PEM的种类包括掺杂改性全氟磺酸型聚合物膜3,6、纯无机质子导体型HT-PEM7、磷酸掺杂型HT-PEM23和膦酸化聚合物膜等17。其中,磷酸掺杂型HT-PEM,由于兼具工作温域宽(130-200 °C)、高温低湿离子电导率高(4 ×10-2- 1 × 10-1S·cm-1)、制备过程简单等优点,被认为是最有实用化前景的高温聚合物电解质膜材料19,23,24,目前已被广泛用来构建实用化的高温聚合物电解质膜电极(HT-MEA)。在电解质膜内,结合磷酸通过酸碱作用锚定在聚合物高分子官能团周围,自由磷酸由于和聚合物分子作用较弱,游离在聚合物膜中构建更广泛的氢键网络,是质子传递的主要方式23,25;而催化层中,在膜电极组装压力和催化层微结构毛细力的作用下,磷酸从电解质膜内迁移进入催化层中,并在电流驱动下在催化层中进行再分布,构建“固-液-气”电化学三相反应界面(TPBs),促进HT-PEMFC性能的提升26,27。然而磷酸作为一种液态电解质,具有水溶和运动的特性28-31。游离的磷酸分子填充在聚合物高分子链周围,大幅减弱了高分子链间的范德华作用力,导致电解质膜的尺寸稳定性的下降32。同时,进入催化层中的磷酸填充在催化层中的孔隙结构中,阻碍了反应气体与铂(Pt)催化剂的接触(质量分数为85%的浓磷酸中氧的溶解度和扩散系数分别为3.3 × 10-4mol·L-1和1.21 × 10-6cm2·s-1,比低温常用的全氟磺酸树脂中分别低一个数量级和4倍)33,增大了催化层内的物质传输电阻19,34-37。此外,HTPEMFC运行过程中,副产物水的生成促使磷酸解离成阴离子并在Pt表面的吸附,降低了Pt催化剂的反应活性38,39;生成的气态水提高了反应气体中水的分压,加快了磷酸的流失40,41。磷酸从电解质膜内迁移进入催化层中导致电解质膜质子电导率的下降并造成催化层的“酸淹”,甚至从MEA中泄露而引起金属板的腐蚀42-44。因此,理解磷酸在膜电极中的分布过程并相应对磷酸在膜电极中的含量和分布进行优化和调控,不仅有利于实现更高的HT-PEMFC性能,而且可以降低磷酸的流失和避免磷酸对电极和电池的影响,为提高HT-PEMFC的稳定性提供可能。
本文在梳理HT-PEMFC膜电极中磷酸分布和迁移过程的基础上,分别对国内外和本课题组在电解质膜及催化层中磷酸调控策略等方面的研究进行综合评述,展望其未来的发展趋势以及进一步提升HT-PEMFC膜电极性能的举措,以期对高性能HT-PEMFC的开发提供指导。
2 HT-PEMFC膜电极中磷酸的分布
在HT-PEMFC中,膜电极的离子电导率主要依赖于磷酸含量。电解质膜中的磷酸在催化层毛细作用力、亲疏水物理特性以及电流的驱动下进行迁移,从而在催化层和电解质膜中进行再分布。下文主要对HT-PEMFC膜电极中磷酸在电解质膜和催化层中的分布状态迁移过程分别进行分析和概述。
2.1 磷酸在高温聚合物电解质膜中的分布
磷酸掺杂高温聚合物电解质膜的制备通常是将聚合物膜浸泡在浓磷酸溶液中,通过改变浸泡温度和浸泡时间而获得不同的磷酸掺杂水平。磷酸通过与聚合物膜中碱性/弱碱性功能基团反应自外而内逐渐完全进入聚合物膜中。因此,在起始较短的时间内有较快的磷酸吸附动力学,并且随着浸泡时间的延长,磷酸进入量趋于稳定,吸附减缓。此外,提高浸泡温度可以降低了聚合物高分子链间的范德华力,不仅可以加快起始磷酸吸附动力学,而且可以增加磷酸在聚合物膜内的掺杂水平。Zeis等45利用共聚焦拉曼光谱研究了不同磷酸掺杂水平下AB-PBI膜中磷酸的分布情况,发现聚合物膜内的含N碱性位点被磷酸质子化过程并非均匀发生的,该过程通常需要数个小时直至磷酸到达膜内每个区域。从图2a可以看出磷酸掺杂的AB-PBI在1611和1572 cm-1出现的峰为苯并咪唑环上C=C/C=N的伸缩振动峰,与磷酸的酸性质子和咪唑环上氮原子间键的形成有关。1572 cm-1处的峰强度随着磷酸掺杂水平的提高而增强,而1611 cm-1处的峰强度保持不变。因此,该两处峰的积分强度可作为AB-PBI膜与磷酸作用程度的描述符,并基于此可得到AB-PBI与磷酸作用程度二维拉曼光谱图45。如图2b,随着浸泡时间由1 h增加至6 h,拉曼强度谱图显示磷酸在膜内的分布更均匀,意味着整个膜基体内含N碱性位点完全被质子化。磷酸在膜内的均匀分布更有利于实现高的质子电导率,而质子在膜中的传递主要依靠于磷酸在膜内构建的动态氢键网络。如图2c所示,对于典型的磷酸掺杂聚苯并咪唑(PBI)高温聚合物电解质膜,其质子传递机理严重依赖于膜内磷酸掺杂水平(ADL),即PBI结构单元和磷酸分子比例22,46-49:(1)没有磷酸掺杂时,质子在PBI咪唑环中的碱性位点间跳跃传递;(2) ADL < 2时,质子主要在咪唑环中的N―H和H2PO4-间跳跃传递;(3) ADL > 2时,锚定在聚合物高分子链周围的结合磷酸与游离磷酸通过氢键作用形成氢键网络,质子通过氢键的不断断裂和形成的动态过程中沿着氢键网络进行传递,该质子传递过程也称为Grotthus质子传递机制。因此,为了增加电解质膜的质子电导率,需要提高膜内的磷酸掺杂水平构建更广泛的质子传递路径。然而,更高的磷酸掺杂量会导致功能基团与磷酸间作用力的弱化,从而增加电解质膜内游离磷酸的含量。在电流的驱动或者水分子与磷酸之间更强的结合能作用下,会使得一部分自由磷酸分子从电解质膜内流失24,50。

图2 (a) AB-PBI膜(黑线)和浸泡6 h(粉色线)的拉曼谱,(b) AB-PBI膜在浓磷酸中浸泡不同时间的二维拉曼光谱图,(c)质子在磷酸掺杂PBI中的传递过程Fig. 2 (a) Raman spectra of pristine (black line) and 6 h doped (pink line) AB-PBI; (b) confocal Raman mapping of AB-PBI membranes with different doping times; (c) chemical structure of PBI, and the proton transfer along acid PBI-acid.
2.2 磷酸在催化层中的分布及动态迁移过程
催化层中的磷酸除了MEA组装前额外添加外,主要是通过电解质膜中过量的磷酸迁移进入。主要包括以下三个过程:①在MEA热压和装配过程中,机械压力会使电解质膜内和表面残留的磷酸溢出进催化层的裂缝中;②然后,在毛细作用力(由催化层孔隙结构和亲疏水性决定)和磷酸表面张力的共同作用下,磷酸在催化层的孔结构中重新分布构建“固-液-气”电化学三相反应界面51,52;③电池运行时,电流驱动少量的H2PO4-(质量分数约占2%-4%)向阳极迁移,在膜内形成的浓度梯度差会驱动磷酸背扩散到阴极侧,阴极处生成的水致使更多的磷酸进入阴极催化层中,该动态过程也促进了磷酸在整个MEA中的再分布24,53-55。Schmidt等56利用同步辐射X射线层析显微术(XTM)揭示电流驱动磷酸迁移过程,发现磷酸在膜电极中的重新分布是一个快速的过程,并且在较短的时间内就会造成阳极催化层的“酸淹”。Kwon等57证明电池运行过程中阴阳极催化层中的磷酸含量占膜电极磷酸总量的~20%,磷酸含量超过该比例会在催化层中引起“酸淹”现象。Büchi等55研究了阳极“酸淹”对H2物质传输的影响,表明阳极即使使用低H2含量反应气体(H2/N2体积比为1 : 9),磷酸在阳极的迁移对HT-PEMFC的输出性能也没有产生明显影响,其原因在于H2的扩散系数比O2大一个数量级。Lehnert等41对比了HT-PEMFC在不同操作条件下性能的衰减情况,发现相比于阳极侧,磷酸在阴极的迁移、累积导致的阴极“酸淹”显著降低了电池性能,同时阴极侧Pt颗粒的出现明显团聚现象,并引起Pt在聚合物膜中的沉积。
电池运行过程中,阴极侧的副产物水会增大反应气体中水的分压,导致阴极侧磷酸含量和解离程度的提高,致使磷酸阴离子物种在Pt表面的吸附以及恶化了阴极催化层的物质传输,进一步影响了氧还原反应(ORR)的进行39,58。如图3不同电势下Pt表面的吸附物种示意图58,当电位低于300 mV时,H原子优先被吸附在Pt催化剂表面。当电极电位上升至300-400 mV范围内时,磷酸物种开始部分取代H原子吸附在Pt催化剂表面。当电极电位进一步升至400-700 mV之间时,只检测到少量的磷酸吸附,作者推测Pt表面仍然全部被磷酸覆盖,而只检测到少量磷酸吸附是由于磷酸分子或阴离子在Pt表面的可移动性以及只有在其他吸附质同时存在的情况下才以有序的方式吸附并变得可检测。电位在700-800 mV范围内观察到含氧物种和磷酸的共吸附。电位继续升至900 mV以上时,Pt表面只有含氧物种被检测到。

图3 (a)不同磷酸物种在Pt表面的覆盖度随电压的变化;(b)不同磷酸物种在Pt表面的吸附随电压变化示意图Fig. 3 (a) Coverage of different adsorbates over cell voltage; (b) cartoon illustrating the situation of adsorbates on the Pt surface at different cell voltages.
此外,燃料电池的运行温度对于磷酸的分布和迁移也有明显的影响。电池运行温度低于120 °C时,电池中液态水的存在加快磷酸流失;在120-180 °C间,温度的提升促进磷酸对于电解质膜的“塑化”作用,电解质膜的力学性能降低,从而导致膜内磷酸流失;而当温度提升至180 °C以上时,磷酸由于脱水形成多聚磷酸或者焦磷酸,降低了电解质膜的电导率24,32,40,41。
综上,磷酸在HT-PEMFC膜电极中的分布与迁移过程如图4所示。电解质膜内自由磷酸构建了质子传递的氢键网络,而过量磷酸的存在会导致膜机械强度的下降。电流驱动下,电解质膜内H2向阳极迁移,与阳极侧反应生成的H+结合提高了阳极侧磷酸的含量,磷酸由于浓度梯度差背扩散至阴极侧,并与阴极反应产物水结合,解离生成H2。该过程促进磷酸在催化层中的再分布,导致催化层的“酸淹”,占据Pt活性位点,增大催化层内的物质传输。此外,磷酸间较弱的氢键作用造成磷酸容易随反应气体中生成的水蒸气一起流失出MEA之外。因此,对磷酸在电解质膜和催化层中的分布行为进行调控和优化是提高HTPEMFC性能和稳定性的关键。

图4 HT-PEMFC膜电极中磷酸的迁移示意图Fig. 4 Migration of phosphoric acid in HT-PEMFC membrane electrode assembly.
3 HT-PEMFC膜电极中磷酸的调控策略
3.1 磷酸在聚合物电解质膜中的调控
HT-MEA中磷酸主要分布在聚合物电解质膜中,高的磷酸掺杂量会导致电解质膜尺寸稳定性的下降以及引发磷酸流失。向聚合物膜中添加无机或有机质子导体59-64,或者在电解质膜内构建磷酸聚集的微观相分离区域65-74,不仅可以减弱磷酸对高分子主链的塑化作用,而且可以优化电解质膜内质子传输环境,从而获得兼具高的质子电导率和机械强度的HT-PEM。此外,还可通过提高电解质膜中官能团与磷酸间的作用力抑制磷酸的流失49,75-78。表1对比了近几年来通过掺杂和分子链结构设计而实现膜内磷酸分布调控与优化的HTPEM的关键参数。

表1 聚合物电解质膜的调控和优化Table 1 The regulations and optimization strategies of polymer electrolyte membranes.
3.1.1 无机/有机质子导体增强电解质膜的质子传递和力学性能
向聚合物内添加无机纳米材料是一种简单有效的改善聚合物电解质膜性能的方法。向磷酸掺杂型HT-PEM聚合物基体材料中添加无机纳米颗粒(可以提升磷酸掺杂水平,同时增加电解质膜的韧性59-61。吉林大学刘佰军等59将SiO2纳米颗粒加入至交联聚苯并咪唑膜网络结构中,使复合膜的磷酸吸附量提高了1.36倍,达到376%。并且该膜在100 °C水蒸气下,6 h后磷酸流失仅为36%,远低于未添加SiO2电解质膜的56%磷酸流失量。Rao等60将高温质子导电功能的α-ZrP纳米粉体添加到ABPBI中得到复合膜,在其添加量为10%时,复合膜的磷酸掺杂水平由每个结构单元吸附3.6个磷酸分子提高至6.5个磷酸分子,180 °C时的质子电导率由39 mS·cm-1提高至46 mS·cm-1,且拉伸强度也提高了约80%。最近,Hooshyari等61将具有质子导电功能的ABO3型钙钛矿材料(SrCe0.9Yb0.1O3-δ)添加至PBI与磺化聚酰亚胺的共混膜中制备复合HTPEM,当钙钛矿纳米颗粒掺杂量为7%时,HT-PEM的磷酸掺杂量最高,达到由每个结构单元吸附14个磷酸分子,此时离子电导率也最高,在180 °C时达到130 mS·cm-1。
近来,本课题组在前期开发的聚醚砜-聚乙烯吡咯烷酮(PES-PVP)高温聚合物电解质膜中添加C3N4纳米片62,同时大幅提升了PES-PVP的离子传导性能和拉伸强度。其原因在于C3N4和磷酸之间存在强的相互作用,质子不仅可以通过磷酸分子之间构建的氢键网络进行传递,还可以通过C3N4和磷酸之间进行传递;因此,质子传递的活化能明显降低。在C3N4添加量为0.5%时,复合膜的质子电导率高达104 mS·cm-1@160 °C,比未添加时提高了36%。最近,我们将磷钨酸掺杂进PES-PVP膜中63,所获得的复合膜的质子电导率也显著提升,在180 °C下电导率高达175 mS·cm-1。核磁共振氢谱测试结果表明,磷钨酸的存在提高了膜内磷酸的解离程度,产生更多自由质子,从而实现电解质膜电导率的提升,并且有望通过调节磷钨酸的含量来降低膜内的磷酸掺杂水平。同时,在我们的工作中也发现有机多元膦酸与磷酸之间也存在强氢键作用,将其添加进PES-PVP膜中在维持磷酸的掺杂量近似一致的条件下(质量分数约170%),有机多元膦酸添加的电解质膜电导率提高了89%(由41 mS·cm-1提高至77 mS·cm-1@180 °C),为实现低磷酸含量HT-PEM的开发提供了可能(未发表工作)。东北大学何荣桓等64将亲水性的碳量子点掺杂进PES-PVP膜中,在与未掺杂膜相近的磷酸含量下,质子电导率达到39 mS·cm-1@180 °C,提高了约35%,而且拉伸强度也提高了16%。
3.1.2 构建磷酸聚集的微观相分离区域
聚合物高分子链上含氮功能基团是吸附磷酸的主要位点,其化学分子结构、在高分子链上的位置以及碱性强弱都对磷酸的掺杂水平及磷酸对聚合物的塑化作用有较大的影响65-70。因此,相关研究者在调节聚合物高分子链功能基团上进行了大量研究。LT-PEMFC使用的聚电解质膜Nafion®具有独特的分子结构,其亲水的磺酸侧链提供了质子传递的通道,疏水的全氟主链支撑了膜的力学性能71-73。受其启发,本课题组对聚合物高分子链进行合理设计,改变磷酸吸附功能基团在高分子链的位置以及个数(图5a),在电解质膜内形成微观相分离区域,既构建了质子传输的离子通道又降低了磷酸分子对高分子主链的塑化作用。通过合成一系列具有不同侧链长度的末端咪唑官能化聚苯醚(PPO-PIm-Cx)聚合物膜,发现随着侧链长度的延长,该膜的磷酸吸附水平逐渐降低,在Fenton试剂中的耐氧化效果也明显提高。当侧链中亚甲基的数量为6时,电解质膜内形成的微观相分离区域尺寸最大为5.56 nm,160 °C时的电导率达到42 mS·cm-1,拉伸强度为12.1 MPa (未发表工作)。为了进一步提高聚合物膜的磷酸吸附水平,本课题组通过卤化反应将含有三个叔胺基团的2,4,6-三(二甲氨基甲基)苯酚(TDAP)和含一个叔胺基团的二甲胺(DMA)分别接枝到聚砜(PSU)主链上65。由于更大的离子团簇的形成,PA掺杂TDAP-PSU膜在与DMA-PSU膜相近的质子电导率下,具有更高的拉伸强度;在相近的拉伸强度下,具有更高的质子电导率。此外,本课题组利用原子转移自由基聚合(ATRP)方法将锚定磷酸的含氮功能基团乙烯基咪唑接枝到PSU分子主链上,获得系列具有不同侧链长度和功能基团数量的聚合物高分子材料66。其侧链结构有利于构建磷酸掺杂HT-PEM的微观相分离结构,侧链结构上的含氮功能基团数量的增加可以提高磷酸的吸附水平形成更大的亲水离子团簇。在拉伸强度保持在7.94 MPa条件下,质子电导率高达127 mS·cm-1@160 °C。
除了对侧链结构的调控,本课题组通过对高分子主链结构的设计,制备了含哌啶疏水芳基骨架的聚芳哌啶类聚合物(PPT)74。经磷酸掺杂后的聚苯并咪唑膜(OPBI)在原子力显微镜(AFM)和小角X射线散射(SAXS)测试结果均未发现微相分离结构的形成。然而,磷酸掺杂的PPT膜,不仅在AFM图像中有明显的相分离区域出现,而且如图5b所示,SAXS曲线也表明在电解质膜内形成了磷酸聚集的离子团簇。因此,虽然该聚合物膜有与OPBI相近的磷酸掺杂水平,但是其在180 °C时的质子电导率却高达96 mS·cm-1,拉伸强度达到12 MPa,远优于OPBI膜。
综上,如图5c所示,通过对聚合物膜的源头设计,进行高分子结构的精细调节,促使在电解质膜内形成微观相分离的磷酸聚集区域(含氮功能基团形成的离子团簇锚定磷酸后为质子的传输提供离子通道,憎水的主链结构构成膜的骨架),降低磷酸分子对高分子主链的影响,从而实现高温膜材料离子导电性能和力学性能的双赢。

图5 (a)聚合物高分子结构设计示意图,(b)磷酸掺杂的聚芳哌啶(PPT)和聚苯并咪唑(OPBI)的SAXS曲线,(c)电解质膜质子传导率和力学性能随磷酸掺杂水平示意图Fig. 5 (a) Schematic diagram of polymer structure design; (b) the SAXS curves of PPT and OPBI membrane with phosphoric acid doping; (c) schematic diagram of proton conductivity and mechanical properties of electrolyte membrane with phosphoric acid doping.
3.1.3 通过季铵基团与磷酸形成离子对抑制磷酸流失
磷酸作为一种中强酸难以完全解离,因而电解质膜内的质子活度远低于磷酸的浓度。电解质膜中过量的磷酸构建了质子传递的氢键网络,然而水分子和磷酸间强的氢键作用又会加速磷酸的流失。以典型的磷酸掺杂型HT-PEM (PA/PBI)为例,PBI高分子链中咪唑功能基团的碱性较弱,导致PBI与磷酸之间产生的分子间作用力PBI···H―H2PO4较小,质子更靠近H2PO4-,致使PBI膜需要更多的碱性位点以及高的磷酸浓度去获得高的质子电导率49,75。Lee等49制备了磷酸掺杂的季胺化的聚苯基高温质子交换膜,其中苄基三甲基铵与磷酸之间分子间作用力为634.7 kJ·mol-1,远大于PBI与磷酸间的72.8 kJ·mol-1以及水分子和磷酸间的52.7 kJ·mol-1。这种强的作用力牢牢地将磷酸锚定在电解质膜内,既提高了磷酸解离程度,使电解质膜拥有较高的质子电导率,又减缓了由于水的吸附带走大量磷酸的过程。因此,该膜在水分压9.7 kPa,80-160 °C的热循环下电压衰减为1.51 mV·h-1,磷酸保留量为60%,远高于同等条件下Celtec®MEA的15%的磷酸保留量。Quartarone等76使用含有碱性功能基团的无机填料掺杂到PBI膜内也达到了相似的效果。Liu等77通过将离子液体填充进一维的硅纳米棒中制备PBI复合膜,在低的磷酸掺杂(ADL = 1.93)下,磷酸保留率达到65%,质子电导率48 mS·cm-1@180 °C。长春工业大学王哲等78将含有季胺功能基团的长烷基链接枝到PBI主链上,聚合物膜的耐氧化稳定性和机械性能均有大幅提升,并且在80 °C和49% RH下,72 h后磷酸保留率达到80%,150 °C时的质子电导率可稳定维持在78 mS·cm-1。
综上,不同的策略均可在一定程度上改善HTPEM的综合性能,但也各种方法也存在一定的局限性。对聚合物分子链进行源头设计而言,调节碱性官能团的种类和数量以及分子链的结构均涉及较为复杂的化学合成反应过程,会增加聚合物电解质膜的成本。并且,聚合物分子链的结构对于聚合物膜也有较大的影响,例如侧链的延长会导致聚合物膜热稳定性的下降,而碱性位点的增多会导致聚合物膜化学稳定性的下降等。在聚合物膜中通过无机-有机掺杂的方式虽可以提高电解质膜质子电导率、力学性能或保酸能力,然而掺杂的方式也会导致所制备出的聚合物膜产生相分离进而影响聚合物膜的化学及力学稳定性,降低了磷酸的掺杂量,进而影响HT-PEMFC运行的长期稳定性。
3.2 催化层中磷酸的调控
催化层中需要磷酸作为电解质构建电化学反应的三相界面,然而过量的磷酸会造成催化层的酸淹,并且毒化Pt催化剂。因此,研究者在通过催化层结构的调节来调控催化层中磷酸的分布以及抑制磷酸对Pt的吸附等方面进行了大量的研究。表2列举了通过催化层结构的设计而调控HT-MEA中磷酸分布的相关进展。

表2 催化层的调控及优化Table 2 The regulations and optimization strategies of catalyst layer.
3.2.1 减少催化层的裂缝
在HT-PEMFC催化层的制备过程中,由于溶剂蒸发时间、粘结剂的含量和制备方式等不同,容易在催化层中形成大小不同的裂缝。Halter等79研究发现德国BASF生产的高温膜电极催化层的裂缝宽度高达85 μm,平均宽度为40 μm。较大的裂缝宽度和密度降低了毛细作用力,磷酸可快速地通过裂缝进入催化层中,因此容易导致催化层的“酸淹”现象80。如图6a所示,磷酸浸满宽度大于60 μm大裂缝的毛细压力为1.5 kPa,随着宽度降至20-60 μm之间,所需毛细压力增至3 kPa,对于宽度小于20 μm的裂缝则需要更大的毛细压力79-81。而催化层的这些物理特性很大程度上受制备方法的影响。Lee等82研究发现,通过刮涂方式制备的催化层存在宽度较大的裂缝,虽然大量磷酸的存在提高了催化层中质子的传递阻力,但同时导致氧的传输阻力增大。如图6b,本课题组利用超声喷涂、丝网印刷、平板刮涂的方式制备了三种具有不同表面形貌以及孔径结构的催化层。对于丝网印刷和平板刮涂制备的催化层,表面裂缝相互连接且比较紧密,裂缝大小分别为20-30和10-15 μm;而对于超声喷涂制备的催化层表面较为平整,裂缝少且相互不连接。在Pt负载量为0.5 mg·cm-2以及相同的操作条件下,通过喷涂方式制备的阴极催化层电池输出功率分别比涂布和丝网印刷制备的阴极催化层高出10%和56%,并且经过加速老化测试后,性能衰减仅为11%,而其他两种分别为26%和43% (未发表工作)。因此,催化层表面的裂缝越小,将有效地减少磷酸进入催化层中以及降低磷酸在催化层中的聚积。
一维纳米纤维材料由于具有良好的韧性和拉伸强度、一定的长径比,能够起到增强基体和抑制裂纹产生的作用83-86。如图6c所示83,我们将一定量的碳纳米管(CNT)和催化剂墨水混合一起制备催化层,当催化层中CNT的质量分数达到2%时,催化层表面的裂缝明显少于未添加CNT的催化层表面,基于该催化层组装的HT-PEMFC最大功率提高了1.5倍,达到673 mW·cm-2。并且HT-PEMFC在电流密度为1.5 A·cm-2,多次启停下展现更好的稳定性。此外,催化层的制备方法也影响了粘结剂在催化层的分布。Mack等86通过AFM测试催化层表面的局部电流分布,发现用平板刮涂制备的催化层表面会有更多PTFE的存在,提高了催化层表面疏磷酸特性,阻碍了磷酸进入催化层内部,从而需要更长时间对MEA进行活化。
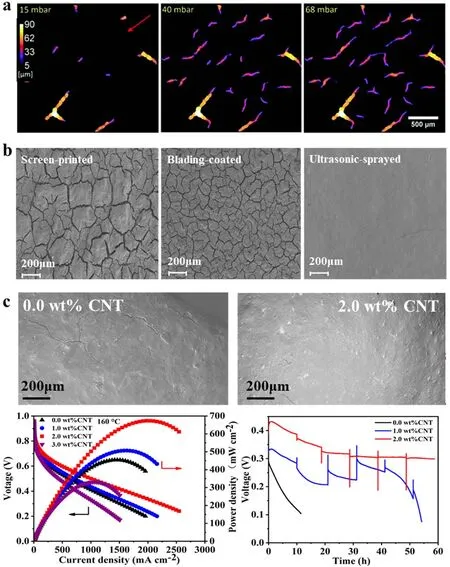
图6 (a)磷酸渗透进催化层的局部厚度分布;(b)催化层不同制备方法的表面形貌;(c)添加CNT前后催化层的表面形貌,基于此催化层组装的HT-PEMFC功率密度和极化曲线以及在160 °C下电流密度为1.5 A·cm-2加速条件下的稳定性Fig. 6 (a) Distribution of the local thickness of phosphoric acid invading the catalyst layer cracks for capillary pressures;(b) top-view SEM images of catalyst layer with different fabrication methods; (c) top-view SEM images of catalyst layer with and without CNT addition and the HT-PEMFC performance and stability at 1.5 A·cm-2 and 160 °C.
3.2.2 调节催化层的亲疏水性
催化层中高的磷酸含量虽然提供了高的质子传导率,但同时阻碍了气体的传输,使用不同的粘结剂以及调控其含量可以改变催化层的亲疏水性,进而调节催化层中磷酸的含量87-89。通常所使用的目前,制备HT-MEA常用的粘结剂有PBI、聚四氟乙烯(PTFE)、聚偏氟乙烯(PVDF)和Nafion®等。使用PBI等氮基团聚合物作为粘结剂可以将电解质膜中的磷酸迁移进催化层中,并且通过与磷酸酸碱结合提供催化层质子传输的路径同时防止硫酸的流失,从而保障了电池运行的稳定性90-92;PTFE等聚合物可以提高催化层的疏水性,降低磷酸的“酸淹”,增大反应气体与Pt催化剂的接触38,88,89;而Nafion®与催化层中的PA结合展现了更高的质子电导率52,93。对于PTFE粘结剂,优化的质量载量为20%,含量过少会降低催化层的疏水性,含量过高会增加催化剂的团聚94,而PBI和Pt催化剂优化的质量比为1 : 491。苏华能等95结合PVDF与磷酸有更好的粘合力而PTFE疏磷酸的特点,制备出一种新型的双催化层结构气体扩散电极,显著提高了HT-PEMFC在大电流区的性能。本课题组与浙江大学和庆刚合作通过微电极电化学装置研究了催化层中粘结剂PVP和磷酸的含量对于O2的物质传输的影响,发现催化层中PVP含量增加和大量磷酸的存在均将导致O2渗透的降低,从而致使HTPEMFC性能的下降89。由于粘结剂的使用可能会降低催化层的孔隙结构影响物质传输,以及阻塞Pt催化剂的活性位点,降低催化剂的活性面积,丹麦科技大学的李庆峰等52进一步发展了无粘结剂的催化层,相比于使用PBI的催化层,基于无粘结剂催化层的HT-PEMFC最高功率提高了1.16倍,并且在900 h的长期运行条件下没有明显衰减。此外,还可以对催化层表面改性,以调节催化层的亲疏水性。Kim等26通过使用Pt脉冲电沉积的方式改变阳极催化层表面的亲磷酸特性,提高了磷酸在阳极催化层的含量及分布,构建了更多有效的三相界面,从而降低了电池的欧姆电阻和电化学反应的电子转移电阻。
3.2.3 调节催化层内部二级孔结构
磷酸通过毛细作用力进入催化层内部参与电化学反应界面的构建。磷酸在催化层内的分布不均不仅会降低Pt利用率,还会增大物质传输阻力,进而降低电池性能和稳定性。如前文所述,降低催化层中裂缝的密度和深度可降低磷酸在裂缝中的蓄积,促进磷酸的均匀分布。此外,催化剂和粘结剂的团聚颗粒大小和粘结剂在催化层中的分布状态会显著影响催化层的微观结构,进而影响磷酸在催化层内的分布。Lobato等96通过使用不同极性的溶剂配制“胶束型”和“溶液型”催化剂墨水。由“胶束型”墨水制备出的催化层其团聚体粒径相对较大,降低了由于PTFE引起的三相界面的电子绝缘影响,展现出较高的Pt利用率。而由“溶液型”墨水制备的催化层团聚体较小,促使了反应气体与Pt的充分接触而降低了催化层内的物质传输。Kim等97提高了PTFE乳液表面活性剂的含量,配制了PTFE均匀分散的催化剂墨水,避免了由粘结剂析出引起的催化剂的团聚,制备出PTFE更均匀分布的催化层,致使催化层电化学活性面积(ECSA)从4.3提高到了12 m2·g-1。由于磷酸主要填充在催化层内的孔隙之间,因此通过调节催化层内部孔径结构,也可促进磷酸在催化层中的重新均匀分布。Cho等94通过改变催化层中PTFE粘结剂的含量,发现在粘结剂质量含量< 20%时,催化层中二级孔结构最优,由此制备出的催化层具有最好的物质传输性能。此外一些造孔剂,如草酸铵、硫酸铵等添加进催化层中,加热挥发后也可以调节催化层中的微孔结构,改善ECSA98。本课题组,通过将CNT添加进催化层中,调节了催化层中的二级孔径结构,ECSA从20.0提高至29.4 m2·g-1;并且在大电流持续放电下,阴极催化层中物质传输的恶化得到缓解。
3.2.4 调控催化层的组成
磷酸是一种液态电解质,进入催化层中的磷酸会在Pt的表面形成一层液膜阻碍反应气体与Pt的接触,并且磷酸解离的阴离子在Pt表面吸附会占据Pt的活性位点,降低整体电化学反应的速率。通过向催化层中添加一些无机固体,改善催化层中的质子传递或与磷酸反应均可降低磷酸对Pt的影响。苏华能等99将质子导体磷酸氢锆(ZHP)添加进催化层中,明显改善了催化层中的质子传导率,降低了电化学反应的电子转移电阻,在电流密度为~0.2 A·cm-2持续放电下,电池的衰减速率仅为~19 μV·h-1。Kim等100研究发现,催化层中添加少量Al2O3可以与磷酸在高温下反应生成Al(H2PO4)3,通过与磷酸的结合而避免了催化层中PA对Pt的影响,同时也抑制了磷酸的流失。最近,苏华能等101将适量席夫碱型共价有机框架材料引入至催化层中,利用其独特的微孔孔径和丰富的含氮官能团结构有效减少了膜电极中磷酸的流失速率,并且电池性能也提升了30%。
增加催化层的厚度均可有效避免磷酸酸淹的同时可降低磷酸对Pt的影响,但增加催化层的厚度通常会造成Pt用量的提高、Pt利用率的降低,增加了膜电极的成本。Li等102,103使用不同Pt含量的催化剂制备Pt载量为~0.1 mg·cm-2的催化层,发现使用质量分数为10%的Pt/C催化剂制备的催化层的膜电极的峰功率密度为321 mW·cm-2@160 °C,是使用质量分数是60%的Pt/C催化剂的1.46倍。并进一步通过使用质量分数5%的Pt/C催化剂实现了膜电极中Pt总用量为0.15 mg·cm-2的超低Pt载量,电池输出功率高达到346 mW·cm-2@180 °C,电池运行稳定性超过2086 h。
与低温相比,在高温和磷酸介质环境下,Pt更容易发生溶解而迁移至电解质膜中形成Pt带,研究者通常将Pt和其他金属(Pd、Ru、V、Cr、Co、Ni、Cu等)104-106合金化去稳定Pt颗粒。然而磷酸阴离子在Pt表面吸附占据了Pt活性位点,影响了电池的性能和运行的稳定性。因此发展非贵金属催化剂去取代Pt用于HT-PEMFC的阴极是解决上述问题和降低HT-PEMFC成本的重要措施之一107,108。如图7所示,本课题组与科廷大学蒋三平等合作109制备了质量载量为7.7% ± 1.3%高金属载量的Fe单原子催化剂,并将其用于HT-PEMFC的阴极中,在230 °C电池的最高输出功率与Pt催化剂相近,达到325 mW·cm-2,在0.5 V持续放电下性能衰减仅为16%低于Pt催化剂的38%。

图7 (a) FeSA-G催化剂的STEM-EDS mapping和(b)AC-STEM图;(c) FeSA-G催化剂和Pt催化剂组装的HT-PEMFC在230 °C下极化曲线Fig. 7 (a) STEM-EDS mapping and (b) AC-STEM images of FeSA-G; (c) the polarization curves of HT-PEMFC based on FeSA-G and Pt/C at 230 °C.
综上,HT-MEA催化层的组成和制备工艺会显著影响HT-PEMFC的输出性能和稳定性。使用超声喷涂工艺可提高催化层表面的平整度减少裂缝的生成,使用疏水的PTFE作为粘结剂构建疏水的催化层,两者均有利于降低磷酸在催化层中的蓄积,减少磷酸流失的途径。但喷涂制备工艺的效率较低,有待进一步提升;憎水粘结剂的使用虽然降低了催化层中磷酸的积累和流失速率,但同时也降低了催化层的离子电导率和Pt催化剂的利用效率。通过在催化层中添加无机纳米材料或者通过改变配制墨水所用溶剂不仅可以改善催化层内部的微孔结构,还可以调节催化层的亲疏水性,进而调节磷酸在催化层的均匀分布,在构建更多有效三相界面的同时降低物质传输电阻,从而提高HTPEMFC的性能和稳定性。目前,非贵金属催化剂对磷酸有较好的耐受性,已初步展示出可在HTPEMFC中应用的前景,但是其电化学活性与Pt催化剂仍然存在较大差距,更长时间的工作稳定性也有待进一步考察。
4 结论和展望
HT-PEMFC由于较高的工作温度而对燃料和空气杂质气体具有更高的耐受能力而成为PEMFC重要的发展方向之一。高温聚合物电解质膜MEA是HT-PEMFC的核心,其中高温聚合物电解质膜的离子电导率和催化层三相界面均直接与磷酸含量相关,但同时高的磷酸含量会致使聚合物电解质膜机械强度低和催化层反应物传输阻力大、磷酸流失等诸多问题。本文详细阐述了磷酸在电解质膜和催化层的分布和迁移过程。膜内结合磷酸通过酸碱作用锚定在聚合物官能团周围,自由磷酸填充在高分子链间构建质子传递的氢键网络,并在MEA组装压力、催化层毛细作用力和电流的驱动下向催化层中迁移及再分布,并部分占据Pt活性位点。针对磷酸降低电解质膜力学强度、酸淹催化层、毒化催化剂和磷酸流失等关键科学问题综述了目前研究者对磷酸调控的研究进展。
对于高温聚合物电解质膜而言,通过有机-无机复合、聚合物高分子链的设计以及提高含氮功能基团与磷酸的作用力等方式在增强电解质膜质子电导率的同时可有效提高磷酸在电解质膜内的结合能力,从而减缓磷酸从电解质膜内的流失。在HT-PEMFC实际运行的工况下,电池的操作温度、水蒸气含量和气体流量的提高也均会加速磷酸的流失。因此,结合HT-PEMFC实际运行的工况去提升高温聚合物电解质膜的保酸能力是后续研究的重点方向之一。
对于催化层而言,通过降低催化层的开裂程度、合理地增加催化层的憎水性和调控微观孔结构均可有效调节磷酸在催化层内的分布,构建更多有效的电化学反应的三相界面,提高Pt催化剂的利用效率,从而降低HT-PEMFC的成本。催化层中合理的微孔结构也可减少磷酸在催化层中的蓄积量,从而降低磷酸的流失速率,提高MEA整体的保酸能力。然而磷酸从电解质膜进入催化层的迁移过程是一个动态的变化过程,后续研究中需要进一步发展表征催化层中磷酸动态迁移的原位表征技术,实时观测磷酸的动态迁移过程,为催化层的理性设计提供依据。
此外,在电解质膜和催化层的接触界面进行界面结构的设计和调控,可以减缓磷酸从电解质膜内迁移进催化层中的过程,促使磷酸均匀进入催化层中。并且可以利用界面层的独特物理化学优势,起到保护催化层的作用,从而降低碳腐蚀和抑制Pt带向电解质膜中的迁移,提高整体HTPEMFC的稳定性。同时,通过对Pt催化剂的改性、合金化或者使用非贵金属催化剂均可降低磷酸对催化剂的毒化作用,提高催化剂的利用率降低HTPEMFC的成本。
从MEA组装的HT-PEMFC电堆或发电装置上看,受质子导体磷酸高温下脱水聚合及更快地蒸发流失的限制,目前HT-PEMFC工作温度为120-180 °C,与甲醇重整温度(260-300 °C)不相匹配。通过新型中高温质子导体的研发,提升电池器件的工作温度使之与甲醇重整器相耦合,可以提升系统的能量转化效率和热利用率。另一方面降低HT-PEMFC的启动温度(在低温下液态水的存在磷酸容易流失以及低温下较低的质子电导率),可以大幅缩短电堆的启动时间,有利于将HT-PEMFC推向实用。因此,具有宽工作温域的高温聚合物电解质膜和膜电极的研发,也是HT-PEMFC未来的重点研究方向之一。
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
陶瓷学报(2021年1期)2021-04-13
国际放射医学核医学杂志(2020年4期)2020-07-27
中国化肥信息(2019年12期)2020-01-16
中国化肥信息(2018年7期)2018-08-23
中国化肥信息(2018年6期)2018-08-23
中成药(2018年5期)2018-06-06
中国化肥信息(2017年7期)2017-12-13
中学生数理化(高中版.高二数学)(2017年1期)2017-04-16
西北工业大学学报(2015年4期)2016-01-19

- 物理化学学报的其它文章
- 有序金属间化合物电催化剂在燃料电池中的应用进展
- Cost-Effective Hydrogen Oxidation Reaction Catalysts for Hydroxide Exchange Membrane Fuel Cells
- Recent Progress in Proton-Exchange Membrane Fuel Cells Based on Metal-Nitrogen-Carbon Catalysts
- 提升燃料电池铂基催化剂稳定性的原理、策略与方法
- Formic Acid Electro-Oxidation Catalyzed by PdNi/Graphene Aerogel
- Enhanced Performance and Durability of High-Temperature Polymer Electrolyte Membrane Fuel Cell by Incorporating Covalent Organic Framework into Catalyst Layer