
碱性介质中氢氧化和析氢反应机理研究现状
2021-09-28 04:50李孟婷郑星群李莉魏子栋
物理化学学报 2021年9期
李孟婷,郑星群,李莉,魏子栋
输配电装备及系统安全与新技术国家重点实验室,洁净能源与资源化工过程重庆市重点实验室,重庆大学化学化工学院,重庆 401331
1 引言
随着全球变暖和能源安全等问题日益突出,可再生能源的开发和使用方面的问题亟待解决1。其中,氢能是重要的能源载体之一,因其能量密度极高,且在能量与物质的转换过程中无有毒气体和二氧化碳的排放,成为满足世界能源需求和减少有害气体排放的理想选择,并在未来能源的可持续发展与应用中扮演着重要角色2。氢气在交通运输系统中,无论是作为内燃机燃料还是燃料电池燃料,都将大大降低对化石燃料的依赖,减少尾气排放。同时,氢气还可应用于工业合成氨、金属切割以及石油工业过程等各个领域。
实现氢能循环利用有两个关键过程,即制氢与消耗氢3。在众多制氢方法中,电解水制氢操作简单,无有害副产物,受到人们的青睐4。其阴极为析氢反应,电极反应为: H2O + e-→ 1/2H2+OH-。燃料电池(fuel cell,FC)5是将氢能转换为电能的方式之一,因不受卡诺循环的限制,能量转换效率高、无污染、燃料不断则发电不停,被列为“12种改变未来的颠覆性技术”之一。氢氧化反应发生在燃料电池阳极,电极反应为:1/2H2+ OH-→H2O + e-。利用电解水技术获得氢气供给燃料电池发电,可真正实现电能-化学能-电能的能量循环,以及从水到氢再到水的物质循环。
HOR和HER都需要电催化剂来降低电极过程的过电势。质子交换膜燃料电池(proton exchange membrane fuel cell,PEMFC)和质子交换膜电解水(proton exchange membrane water electrolysis,PEMWE)技术中主要采用铂族贵金属作为HOR/HER催化剂。这些贵金属催化剂虽可极大的降低HOR/HER过电势,且具有较理想的动力学反应速率6,但是,它们在地球上有限的储量和昂贵的价格大大增加了燃料电池与电解水制氢的成本。此外,酸性介质对催化剂的腐蚀对其使用寿命提出了挑战,进一步限制了其商业化发展和大规模应用。碱性膜燃料电池(alkaline membrane fuel cell,AMFC)和碱性电解水(alkaline water electrolysis,AWE)可使用地球上储量丰富的非贵金属材料作为催化剂7-10,从成本上更具有商业可行性。然而,在碱性条件下,即使是使用活性最高的Pt基催化剂,其HOR/HER动力学也比酸性条件下慢2至3个数量级11,严重阻碍了低温碱性能源转化器件的发展。为了推动碱性介质下燃料电池与电解水制氢技术的发展,科研工作者们致力于解析碱性介质中的HOR/HER机理12,13,探究碱性与酸性电解质中HOR/HER活性差异的根本原因,以期设计、筛选、制备出具有高活性和稳定性的HOR/HER非贵金属催化剂。
现已有许多文献对HOR/HER催化剂的发展14,15、设计策略16,17、制备方法18,以及HOR/HER机理研究13,19,20进行了相关总结和介绍。在此,本文重点对近年来碱性介质中HOR/HER机理的相关解释与推论,以及各观点存在的争议进行总结和讨论;并从理论计算的角度,介绍了目前电化学界面的理论模拟方法的发展及其在HOR/HER研究中的应用。
2 碱性HOR/HER机理
如图1所示,铂基催化剂在碱性电解质中的HOR/HER活性至少比在酸性介质中低2至3个数量级11。换句话说,要使AMFC达到与PEMFC相同的电流密度,需使用更多的阳极催化剂或增加阳极过电势,其大大降低了AMFC的功率密度、提高了制造和使用成本。由此,对碱性HOR/HER的反应机理、碱性HOR/HER动力学缓慢的原因、碱性HOR/HER催化剂的活性描述符以及低成本高活性HOR/HER催化剂尤其是非贵金属催化剂的研究成为科研工作者们关注的焦点。

图1 (a) 353 K时,PEMFC中Pt/C的HOR/HER交换电流密度,与在0.1 mol·L-1 KOH溶液中外推到353 K所得的交换电流密度值的对比;(b) KOH溶液中,不同质量活性的Pt/C的HOR过电势(黑色实现和灰色虚线);353 K时,Pt/C在PEMFC中测得的质量活性(红线) 11Fig. 1 (a) HOR/HER specific exchange current density of Pt/C in a PEMFC at 353 K compared to that obtained in 0.1 mol·L-1 KOH and extrapolated to 353 K. (b) HOR overpotential of Pt/C with different mass activity in KOH (black solid lines and gray dashed lines); this is compared with mass activities measured on Pt/C in a PEMFC at 353 K (red lines) 11.
由于电催化体系的复杂性,碱性介质中的HOR/HER动力学比酸性介质中慢的本质原因仍不明确,并且碱性介质中HOR/HER的反应机理也存在一定争议。特别是,碱性介质中HOR/HER中的Heyrovskey和Volmer步骤均涉及到了OH-的参与,OH-的来源以及其如何与H作用影响反应机理与反应动力学成为争论的焦点。近年来,碱性介质中HOR/HER机理的解释主要包括以下几种:双功能机理、氢结合能(HBE)理论和电子效应。
如图2a所示,HER与HOR的双功能机理具有不同的描述。HER机理涉及到水的解离,以及Had的复合,其中水的解离是HER的关键步骤之一。HER的双功能机理认为在催化剂表面引入促进水解离的物质可以提高催化剂的HER活性。HOR的双功能机理认为:HOR机理是吸附态的Had与吸附态的OHad反应,生成水后脱附,催化剂的亲氧性与HOR活性之间存在正相关关系,即在催化剂表面引入容易形成OHad的物质,可以提高HOR活性。HBE理论认为Had的吸脱附难易直接决定和影响着催化剂的HOR/HER活性。如图2b所示,HOR的反应路径可看作是Had与溶液中的OH-结合生成H2O,Had是反应过程中唯一的关键物种,HER则是其逆过程。因此,Had在催化剂表面的结合能——HBE成为影响HOR/HER动力学的关键和主要因素。电子效应则重点关注HOR/HER可能的中间物种以及催化剂组分对活性位点电子结构的影响,其认为OH、H2O等吸附物种、催化剂各组分如金属、氧化物等会对催化剂表界面的电子结构产生影响,从而改变Had吸附能,以此来调变HOR/HER机理和反应动力学。
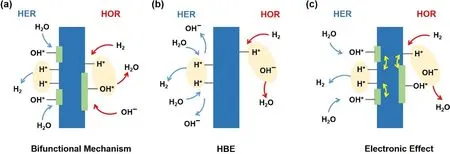
图2 碱性HOR/HER机理示意图Fig. 2 Diagram of alkaline HOR/HER mechanisms.
对于上述碱性介质中HOR/HER机理的解释,众多的研究者都持有各自不同的见解,也在一定程度上通过实验印证了各自的观点,但是迄今还没有统一的理论用于解释所有的实验现象。该部分在简单介绍HOR/HER基元步骤的基础上,着重总结并分析了近年来碱性介质中HOR/HER机理的研究成果和存在的争议。
2.1 HOR/HER基元步骤
HOR/HER主要包括Tafel,Heyrovsky和Volmer三个基元步骤,酸性介质中的反应机理如下:

式中,*代表催化剂上的活性位,Had代表吸附态的H。
碱性或中性介质中,因质子不直接参与反应,基元步骤Heyrovsky和Volmer为:

分析电化学极化中过电位与电流密度之间的关系是考察电化学动力学特征,以及反应决速步(rate-determining step,RDS)的有效方法之一,通常,可直接通过分析电化学极化曲线的Tafel斜率(Tafel slope,TS)来判断HOR/HER遵循的路径或反应机理。Shinagawa等21通过微观动力学分析,提出了HOR/HER中各决速步的理论Tafel斜率。依据反应决速步的不同,HOR/HER有四种不同的反应机理 : Tafel(RDS)-Volmer、 Tafel-Volmer(RDS)、Heyrovsky(RDS)-Volmer和Heyrovsky-Volmer(RDS)。表1汇总了HOR/HER中各决速步的理论Tafel斜率和对应机理的实验Tafel斜率13。

表1 HOR/HER中各决速步的理论Tafel斜率、HOR/HER机理与对应Tafel斜率(298 K)Table 1 The theoretical Tafel slope of the rate-determining step in HOR/HER, mechanism of HOR/HER and corresponding Tafel slope (298 K).
对Tafel(RDS)-Volmer机理,因为Tafel步骤是一个不涉及电子转移的化学反应,反应动力学不遵循Butler-Volmer方程,因此HOR/HER的Tafel斜率均为30 mV·dec-1。例如,John等22采用化学气相沉积技术合成的RuxPty合金的Tafel斜率为~30 mV·dec-1, 确 定 Tafel步 为 RDS , 反 应 遵 循Tafel(RDS)-Volmer机理。碱性介质中的Rh电极23和Ir电极24上的HOR也遵循该机理。此外,有部分研究者25在低过电势下测得Pt上发生HER的Tafel斜率约为30 mV·dec-1,认为RDS为Tafel步。该结论存在争议,因为只有在强极化状态下(理论过电势高于118 mV,通常实际过电势高于60 mV26),逆反应速率可以忽略不计时,Tafel斜率值才能用于判断反应机理。
关于Tafel-Volmer(RDS)机理,在298 K时,HOR和HER具有相同的Tafel斜率值(118 mV·dec-1),所得的Butler-Volmer图形具有对称性,此现象在酸性和碱性介质中的Pt和一些Pt族金属催化剂上的都可以观察到。例如,Zheng等27发现pH从0-13范围内四种负载型铂族催化剂(Pt/C、Ir/C、Pd/C和Ru/C)的反应机理均为Tafel-Volmer(RDS)。值得注意的是,在实验测量过程中,由于酸性介质中Pt的HOR动力学非常迅速,传统的旋转圆盘电极(rotating disk electrode,RDE)中H2在液相的传质比在气相中慢5个数量级28,导致真实的反应动力学电流受H2的液相传质极限电流所掩盖,最终测得的Tafel斜率值往往小于118 mV·dec-1。因此,在酸性介质中Pt催化剂的HOR动力学电流必须通过微电极、气体扩散电极、H2-pump法等不受传质限制的方法来测定29-31。
在 Heyrovsky(RDS)-Volmer和 Heyrovsky-Volmer(RDS)反应机理中,Butler-Volmer图形具有非对称性,这种非对称性来源于电子在RDS的前一步或RDS的后一步中参与反应。例如,Sheng等11指出,KOH溶液中Pt催化剂上的HOR遵循Heyrovsky-Volmer机理,且决速步为Heyrovsky步。因为只有Heyrovsky或Volmer步骤作为RDS才能得到与Butler-Volmer方程一致的动力学表达式,并由此排除了以Tafel步为RDS的可能性。密度泛函理论(density functional theory,DFT)计算发现Heyrovsky步的活化能大于Volmer步,同时基于从头算(ab initio)的微观动力学模拟(micro-kinetic modeling)也 发 现 Pt(111)上 HER遵 循Heyrovsky(RDS)-Volmer机理。
值得注意的是,HER中Tafel(RDS)-Volmer和Heyrovsky(RDS)-Volmer机理的Tafel斜率分别为30和39 mV·dec-1,数值非常接近。在实验中仅通过测量Tafel斜率很难得出唯一明确的反应机理。HOR的 Tafel(RDS)-Volmer和 Heyrovsky-Volmer(RDS)机理中也存在类似的情况。另外,催化剂的Tafel斜率还会受pH和电势等外界因素的影响,导致同种催化剂在不同的反应环境中,可能具有不同的Tafel斜率值21。以Pt催化剂为例,典型的Pt/C电催化剂在0.5 mol·L-1的H2SO4溶液中的Tafel斜率为30 mV·dec-132,在PEMFC环境下的Tafel斜率为120 mV·dec-133,而在0.5 mol·L-1的NaOH溶液中Tafel斜率为125 mV·dec-134。另外,在过电势较低时,Pt/C的Tafel斜率的值为30 mV·dec-1,而过电势较高时,其值为120 mV·dec-1。因此,采用Tafel斜率分析反应机理时,必须注明电解质,并选择适合的电极电势范围。此外,可以考虑引入反应速率等数据来进行更加深入的分析。例如,Montero等23,24用反应中间体的Frumkin吸附模型,将碱性介质中Ir和Rh的HOR极化曲线分别按Heyrovsky-Volmer路径和Tafel-Volmer路径的动力学表达式进行拟合,得到了各个基元步骤的反应速率为vHeyrovsky< vTafel< vVolmer,且Heyrovsky的反应速率比Tafel的反应速率低7个数量级,这就意味着碱性介质中Ir和Rh电极上HOR实际上不会沿Heyrovsky-Volmer路径进行,而是遵循Tafel-Volmer机理,且Tafel为RDS。
2.2 HOR/HER双功能机理
2.2.1 HER的双功能机理
Markovic团队35,36和Li等37发现通过表面沉积的方法在Pt表面(包括Pt(111)、多晶Pt和Pt/C纳米颗粒)沉积Ru和Ni后可以提高Pt上的HER速率;并且在Pt(111)表面添加Ni(OH)2后,虽然表面可用的Pt位点减少了,但是相比于纯Pt(111),复合电极的HER活性却提高了几倍,证实易氧化的金属和Ni(OH)2在促进HER活性中起到了重要作用。据此,Markovic等38提出了HER的双功能机理:Ni(OH)2促进水的解离,解离产物OHad停留在亲氧位点上,而Had则吸附在邻近的Pt位点上,最终两个Had复合生成H2,如图3a所示。其中H2O分子的解离过程需要O原子与Ni(OH)2、H原子与Ni(OH)2边界处的Pt原子的共同作用来实现。实验还发现,在溶液中加入Li+后,催化剂的HER活性会进一步提高。HER的双功能机理将其解释为:水合阳离子AC+与Ni(OH)2之间的非共价相互作用可稳定AC+,AC+进一步与H2O分子作用,改变H2O的取向以及氧化物与H2O相互作用的强度,促进水的解离,从而促进HER动力学。
如图3b所示,Markovic等39还分别比较了不同过渡金属在酸性和碱性介质中的HER活性。他们发现铂族金属Pt和Ir在酸性溶液中的HER活性没有明显差异,但是在碱性溶液中却存在一定差异。当在金属表面负载Ni(OH)2后,Pt和Ir在碱性溶液中HER活性的差异消失,从而印证了H2O的解离在HER反应中的重要性。此外,3d过渡金属V、Ti、Ni材料的表面易被氧化物覆盖,表面性质难以定义,但是表面引入Ni(OH)2后,HER活性得到明显提升,进一步证明了含氧物种对H2O解离的促进作用。因此他们提出,碱性条件下水解离困难是造成HER动力学缓慢的主要原因。

图3 (a) Ni(OH)2/Pt(111)上的HER示意图38;(b)电流密度为5 mA·cm-2时,过渡金属表面在0.1 mol·L-1 HClO4和0.1 mol·L-1 KOH中的HER过电势,以及Ni(OH)2修饰的过渡金属表面在0.1 mol·L-1 KOH中的HER过电势Fig. 3 (a) Schematic representation of the HER on Ni(OH)2/Pt(111); (b) comparison between activities for the HER,expressed as overpotential required for a 5 mA·cm-2 current density, in 0.1 mol·L-1 HClO4 and 0.1 mol·L-1 KOH for both bare metal surfaces and Ni(OH)2-modifed surfaces.
为了证明上述结论,Markovic团队36将3d过渡金属的氢氧化物负载在Pt(111)表面上,构成M2+δ(OH)2-δ/Pt(111)催化剂,通过固定Pt-Had吸附能相关的描述符,将HER作为“伪”单变量函数来处理,即通过控制OHad-M2+δ键强来改变催化剂的HER活性。电化学测试发现,各过渡金属氧化物上OHad-M2+δ键强顺序为:Ni < Co < Fe < Mn,而HER的活性顺序为:Mn < Fe < Co < Ni。由此认为,OHad-M2+δ键强度与材料的HER活性呈负相关关系,即:在碱性溶液中,HER动力学是由水的解离步骤和水解离产物与催化剂表面相互作用之间的微妙平衡决定的。
基于HER的双功能机理,Peng等40设计了Ni(OH)2和过渡金属改性的Ni(OH)2/NiMoPOx复合催化剂,当过电势超过65 mV (vs RHE,下文中未标明电势的参考态时,则表明参考态为RHE)时,其碱性HER活性高于Pt/C。实验数据和DFT计算结果表明,该催化剂HER活性的提升源于Ni(OH)2对水解离的促进作用以及NiMoP表面O的引入对Had吸脱附平衡的调节。
尽管如此,Juarez7和Durst41等对HER的双功能机理提出了质疑。他们认为在Pt表面,H2O是一种非常快的Had供体,水的解离不应是决定HER动力学的关键步骤,并且在酸性和碱性溶液中,由于两种不同的Had的来源会导致HOR/HER速率随着pH的变化发生急剧的改变,而不是实验上所观察到的逐渐变化。Koper等42则发现催化剂的零电荷电势(potential of zero free charge,pzfc)会随着pH的变化而发生移动,导致在碱性介质中HER电势远离pzfc。这说明,在碱性介质中,HER电势处的界面水与界面电场作用增强,H+/OH-等难以穿过界面双电层区域,使得界面水的重组能更高,氢的吸附速率减小,最终导致Volmer步骤具有更高的能垒,即在碱性HER过程中,水的解离不是速度控制步骤。据此,关于HER的双功能机理的更多细节还需进一步完善和深入研究。
2.2.2 HOR的双功能机理
Angerstein-Kozlowska和Conway43曾明确表明Au(111)表面在电势高于0.6 V时才具有HOR催化活性,但这一结果一直没有得到合理的解释。Markovic等35根据如图4a所示Au(111)表面的CV和LSV曲线发现HOR的起始电势与OHad的吸附电势一致,提出Au(111)上的HOR受OHad的电势依赖性吸附控制。由此推测出,OHad在碱性HOR中起着重要作用。此外,他们认为碱性介质中Ir的HOR活性明显高于Pt也可能是由于金属Ir的亲氧性比Pt更高,吸附在Ir表面的OHad可以促进表面氢中间体Had的解吸,从而提高Ir的HOR活性。通过比较Pt0.1Ru0.9和Pt0.5Ru0.5也证实了OHad的作用,因为前者含有更多亲氧性的Ru,导致在碱性环境中Pt0.1Ru0.9的HOR活性是Pt0.5Ru0.5的两倍。据此,Markovic等将Ir和PtRu在碱性介质中的HOR活性与催化剂的亲氧性联系起来,认为OHad是HOR过程中的关键物种,碱性介质中的HOR遵循双功能机理:

图4 (a) Au(111)表面的HOR极化曲线(黑色)和CVs曲线(灰红虚线);(b) 0.1 mol·L-1 KOH溶液中分别滴加0、3、6和9 mL 5 mmol·L-1 RuCl3溶液时Pt/C和Pt1Ru1/C的HOR极化曲线;(c) Ar饱和的KOH溶液中加入不同量的RuCl3时Pt/C的CO溶出伏安曲线;(d) Pd/Ni的结构示意图(左),Pd/Ni表面的双功能催化作用(右)Fig. 4 (a) HOR polarization curves (black) and CVs (dashed grey and red) for Au(111); (b) HOR polarization curves of Pt/C with 0, 3, 6 and 9 mL doped 5 mmol·L-1 RuCl3 and Pt1Ru1/C collected in 0.1 mol·L-1 KOH electrolyte; (c) CO stripping voltammograms collected for Pt/C with various amount of deposited RuCl3 in Ar-saturated 0.1 mol·L-1 KOH; (d) schematic diagram of Pd/Ni structure (left), and a zoom-in to show the bifunctional catalytic effect of the Pd/Ni surface (right).

在此HOR过程中,OHad的形成促进了Volmer步骤的发生。该HOR的双功能机理的关键在于催化剂的表面同时形成Had和OHad,二者反应生成H2Oad后脱附。Li等37观察到在溶液中加入RuCl3可提高Pt/C的HOR活性(如图4b),证实在催化剂表面引入易形成OHad的物质,可以提高HOR活性。
进一步证明HOR双功能机理的一个关键是要提供催化剂表面在HOR电势范围内存在OHad的直接实验证据。Li等37认为在常规的CO溶出实验中,Pt/C的CO溶出曲线上,电势低于0.4 V时不存在CO溶出峰并不代表在HOR反应过程中0.4 V以下不存在OHad。其原因是在HOR条件下,体系中催化剂表面不存在可供CO分子与OH竞争吸附的活性位。如图4c所示,为了排除CO占据活性位的可能,通过CO溶出实验了解0.4 V以下是否存在OHad,作者巧妙地在Pt/C的CO溶出实验过程中向体系加入RuCl3溶液。随着RuCl3量的增加,Pt/C在0.7 V处的CO溶出峰强度逐渐减弱,表明Pt位点数减少;而0.05 V左右的CO溶出峰强度增大,意味着相邻位点的Ru形成OHad促进CO氧化。该实验证明了在Hupd电势区域存在OHad,且对HOR动力学具有促进作用,为HOR的双功能机理提供了证据。然而没有RuCl3时,Pt表面能否在0.4 V下形成OHad,加入RuCl3对Pt/C催化剂表面是否存在其他影响,如活性位的占据、几何、电子结构等的变化目前仍不清楚。
以HOR的双功能机理为依据,许多研究者将亲氧材料复合到催化剂中,并实现了HOR催化活性的提升。如,Alesker等44使用混合了Pd和Ni的纳米颗粒(NPs)作为AMFC的HOR电催化剂(如图4d),得到的峰值功率密度为0.40 W·cm-2,相比于纯Pd作为阳极催化剂时的0.18 W·cm-2,活性有了很大的提升。该研究认为,在不考虑合金的形成以及电子效应的情况下,NPs中Ni的亲氧作用是大幅度提升HOR活性的关键。Alia等45合成了一种Pt包覆的Cu纳米线(Pt/CuNWs)作为HOR催化剂,在碱性介质中,其面积比活性和质量比活性分别是Pt/C的3.5倍和1.9倍。该催化剂活性的提升可归因于Cu基底和表面Cu对Pt电子结构的调节,以及催化剂表面亲氧性的Cu促进了OH的吸附。
然而,Ramaswamy等46提出,双功能机理并不适用于所有催化剂,不同的过渡金属上HOR可能遵循不同的机理。(1)在单金属Pt/C上,HOR的高过电势来源于催化剂表面的Had与OH-作用生成水,此过程需要更高的活化能;(2)在双金属PtRu/C表面,可在相对较低的电势下形成Ru-Hupd…OHad过渡态,Pt-Had与该过渡态发生反应,可在低电势下加速产物H2O的形成;(3)在Pt与Nb、Ni、Cu、Au等过渡金属形成的合金表面,在高pH条件下不是形成Hupd而是一层氧化物,此时才涉及到上述的HOR双功能机理,即Pt-Had与过渡金属氧化物上形成的OHad直接发生反应。此外,Liu等47通过DFT计算发现,在0 V时,OH-比OHad更稳定,OH-更易参与反应;而在0.9 V时,OHad具有更好的稳定性,此时OHad更可能参与反应,HOR遵循双功能机理。由此可见,电势以及催化剂结构、组分都可能对HOR机理产生影响。对在HOR电势范围内不能形成OHad的催化剂,双功能机理似乎不能用于解释其碱性HOR机理和活性变化,由此难以统一采用双功能机理来指导催化剂活性的提升。
2.3 HBE理论
对单金属催化HOR活性的研究,如Durst等41发现碱性条件下的HOR/HER的活性顺序为:Pt/C <Ir/C < Pd/C,与催化剂的亲氧性无关,由此认为双功能机理无法解释上述金属的HOR活性。针对这一问题,Sheng等48提出了HBE理论,该理论认为碱性介质中HOR路径中Volmer步的反应机理为:Had+ OH-→ H2O + e-+ *,即催化剂表面吸附态的Had与溶液中的OH-作用生成水,HER则是逆过程。这种情况下,与酸性介质相同,Had成为HOR/HER的唯一中间吸附物种,最优的HBE可保持碱性介质中Had中间体的吸附和脱附平衡。进一步,作者建立了碱性介质中HBE和HER交换电流密度之间的火山关系图48;通过考察Pt、Ir、Pd、Rh等过渡金属的欠电势沉积氢(Hupd)的峰位置(Epeak)与HOE/HER活性之间的相关性,发现了Epeak与氢的吸附强度EM-H之间的线性相关关系,从而证明了HBE理论27,49的普适性。据此,他们提出HBE可以作为碱性HOR/HER活性的描述符,可通过改变催化剂表面性质来调节HBE值得到最佳的HOR/HER活性。
然而,实验和理论研究均有证据表明Had在催化剂表面的吸附自由能(ΔGH)对pH依赖性可忽略不计50,51。在外加电场下,因为水的偶极与电极之间存在相互作用,仅水在催化剂表面的吸附自由能(ΔGH2O)才对pH具有依赖性。在此基础上,Zheng等52重新定义了氢吸附的表观吉布斯自由能ΔGH,app(the apparent Gibbs free energy of hydrogen adsorption):ΔGH,app= ΔGH- ΔGH2O。其中ΔGH,app可与实验测得的Hupd脱附峰位置直接相关(ΔGH,app= -FEpeak)。从图5a中的Hupd的峰位置可以看出,随着pH的增加,水在Pt(110)和Pt(100)表面的吸附会逐渐减弱从而使ΔGH,app减小,意味着H的表观吸附强度增大。由此,可采用ΔGH,app来解释HOR/HER的pH效应,即:电解质溶液的pH增大导致水的吸附减弱,从而促使氢吸附的表观吉布斯自由能变负,H的表观吸附增强,最终使催化剂的HOR/HER活性降低。Cheng等53通过DFT计算证实了pH增大可减弱水的吸附。他们利用电极电势与pH值间的转换公式,发现当U从+0.29 V变化到-0.46 V (相当于U = 0.3 V,pH从0.2到12.8),带负电荷的Pt(100)逐渐排斥水的吸附。如果假定反应能差与反应能垒之间存在线性关系,根据Arrhenius方程,粗略计算可得pH = 12.8时的HOR活性比pH = 0.2时下降153倍。因此他们认为水的吸附强弱变化是HOR/HER动力学对pH具有依赖性的主要原因。但是该计算对pH的模拟是通过电势与pH之间的公式转化关系实现,真实条件下的不同pH值对水吸附作用的影响仍不清楚。特别是碱性介质中催化剂表界面为何会减弱水的吸附,而弱化水分子为什么可以增强表观H的吸附强度?另外,有研究提出水的重组能的变化以及质子给体从酸性介质中H3O+变为碱性介质中的H2O也会导致HOR/HER对pH依赖41,42,那么上述变化是否也是造成H表观吸附强度变化的因素?显然,电极/溶液界面间溶剂与HOR/HER反应物、产物间的相互作用,电场的影响,以及所引发的微观机制和量化图像仍不清楚,有待进一步深入研究。

图5 (a)在不同pH的电解质中,Pt的稳态CVs;(b)根据H1a峰(电势为0.13 V)的面积归一化所得交换电流密度(i0)与t-ECSA的关系;Ir/C的HOR/HER的t-ECSA归一化所得i0与t-ECSA的关系Fig. 5 (a) Steady state CVs of Pt collected in different pH electrolytes; (b) exchange current densities (i0)normalized to surface area of H1a (peak potential 0.13 V) as a function of t-ECSA; i0 of HOR/HER on Ir/C samples normalized to t-ECSA as a function of t-ECSA.
Zheng等54通过实验进一步佐证了HBE理论。他们合成了颗粒大小不一的5种催化剂,其电化学活性表面积顺序为:Ir/C > Ir/C-300 > Ir/C-500 >Ir/C-600 > Ir/C-800,在0.1 mol·L-1KOH溶液中,HOR/HER活性为:Ir/C-800 < Ir/C-600 < Ir/C-500 <Ir/C-300 < Ir/C,这与总的电化学活性表面积顺序恰恰相反。作者将所得的Hupd解吸峰分解为4个峰,分别代表不同的H吸附位点。在四类位点中,H1a位点的HBE值最小,对应的Hupd最弱,且H1a位点的HOR/HER活性与电化学活性表面积无关,如图5b所示,证明具有最小HBE值的位点是催化HOR/HER最优活性位点。HBE值最小的位点与HOR/HER活性之间的相关性为HBE理论提供了证据。
其他研究者也直接或间接地证明了HBE理论,同时也存在部分争议。例如,Lu等55用Pt基纳米颗粒作为催化模型来确认OHad的影响。通过比较Had结合能基本相同但OHad结合能相差较大的PtNi纳米颗粒和酸洗后的PtNi纳米颗粒的HOR活性,发现碱性电解质中OHad不是影响HOR活性的主要因素,HBE可以作为碱性HOR的活性描述符。然而,有研究者通过碱性介质中Ni的CVs特征峰,检测到Pt表面存在金属Ni56,由此认为Lu等的实验并不能证明经酸浸渍处理后的PtNi/C纳米颗粒表面不存在Ni,其结果还需进一步确认。此外,近期有研究者们发现,RuPt/C和NiPt/C等核壳结构的纳米颗粒比纯Pt所构成的Pt/C纳米颗粒具有更好的HOR/HER活性55,57,且认为是Ru或Ni引入的应力效应和配位效应削弱了Pt-Had之间的相互作用,导致催化剂活性的提升。由于双功能机理在解释HOR/HER活性的提升方面需要催化剂表面存在第二种物质,故他们认为该现象可以用HBE理论来解释。然而,在电化学反应条件下,Ru或Ni很可能会迁移到Pt的表面58,因此RuPt/C和NiPt/C等核壳结构的纳米颗粒是否是干净的Pt表面仍存疑。
总之,尽管HBE理论在很大程度上得到了人们的认可,但是仍存在一些其无法解释的实验现象56。例如,增加碱金属阳离子M+的浓度,可以削弱Pt-H2Oad相互作用59,从而增加HOR/HER活性。并且这种效应只存在于Pt的阶梯型表面(高指数晶面)催化的HER中,对HOR速率无影响,同时对Pt(111)等低指数晶面催化的HOR/HER也没有显著影响60。另外,所有Pt表面的HOR/HER速率都随pH的改变而改变,其中Pt的阶梯型表面的Epeak依赖于pH值,而Pt(111)表面的Epeak却不依赖于pH值42。这些现象表明,HOR/HER速率与Epeak之间的相关关系存在特例。简言之,就阳离子效应和Hupd的pH效应而言,Pt(111)表面和Pt的高指数晶面上H的吸附具有完全不同的行为,但是二者的HOR/HER速率随pH的变化却一致。由此可见,将HBE与Hupd峰位置联系起来的HBE理论还需进一步完善,其是否存在另一因素影响着HOR/HER的动力学。
2.4 HOR/HER电子效应
电子效应重点关注催化剂表界面电子结构的变化,其认为催化剂催化HOR/HER时,表界面的OH-/OHad不仅直接与Had作用反应,还可通过影响催化剂的电子结构而影响催化活性。Wang等61通过CO溶出实验发现在0.1 mol·L-1H2SO4溶液中,Pt/C在0.85 V处出现唯一的CO溶出峰,在与Ru合金化(PtRu/C)以后,CO溶出峰负移了0.3 V,表明Ru在酸性环境中的确加速了OHad的形成(图6a,b)。但是在0.1 mol·L-1KOH溶液中,相比于PtRu/C,Pt/C在0.2 V附近出现多个CO溶出峰,且峰电势负移了0.35 V,表明在碱性环境中,Pt表面比PtRu表面更容易产生OHad。由此,不能采用HOR的双功能机理解释为什么在碱性介质中PtRu/C较Pt/C具有更优的HOR活性。此外,作者还发现在碱性介质中,Pt/C的CV曲线中以强Had峰为主,而PtRu/C以弱Had峰为主,且Pt3Ru上的Ru位点被OHad占据后,Pt-Had键会进一步减弱。他们将这种现象归为电子效应,即Ru与Ru上吸附的OHad会调节Pt的电子结构,导致Pt对Had的吸附减弱,从而提高催化剂的碱性HOR活性。
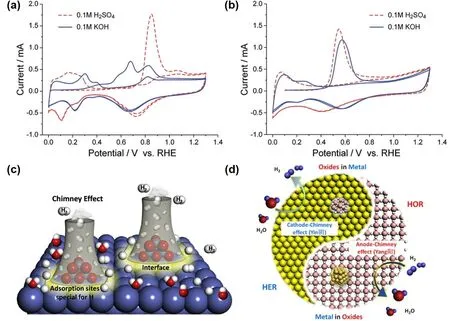
图6 (a) Pt/C和(b) PtRu/C在0.1 mol·L-1 H2SO4和0.1 mol·L-1 KOH溶液中的CO溶出曲线;(c) HER的阴极“烟囱效应”原理图;(d) HOR的阳极“烟囱效应”原理图Fig. 6 CO tripping on Pt/C (a) and PtRu/C (b) in 0.1 mol·L-1 H2SO4 and 0.1 mol·L-1 KOH solutions; (c) schematic diagram of the cathode-chimney effect for the HER; (d) schematic diagram of anode-chimney effect for the HOR.
上述电子效应实际上广泛地存在于复合催化剂中。Peng等62发现将金属氧化物负载在金属上时,如RuO2/Ni、NiO/Ni等属氧化物/金属复合催化剂界面间的电荷转移,可导致金属氧化物/金属界面不吸附OHad和H2Oad,只吸附Had,且界面效应可以调节Had的吸附达到最优值,使其更容易脱附形成H2,大大提高催化剂的HER活性。如同在“金属氧化物/金属”界面周围形成H2快速抽提的“烟囱”,称之为金属氧化物/金属界面对HER的“烟囱效应”(图6c)。HOR在复合催化剂中也存在类似的“烟囱效应”(图6d)63。将Ru簇负载于TiO2表面时,Ru-TiO2通过界面形成的Ru-O键调节界面Ru簇的氧化程度,界面间的电子转移抑制了HOR过程中Ru簇的氧化,导致OHad和H2Oad在Ru位点上的吸附减弱,释放出更多的H吸附活性位,使复合催化剂的HOR质量活性比Ru/C和PtRu/C分别高17.5和1.5倍。此外,调节金属氧化物-金属间的成键类型,也可以有效调节催化剂表界面的电子结构,同步实现催化剂的高HOR活性、抗氧化性以及抗中毒特性。Zhou等64利用“晶格限域”合成了具有大量Ru-Ti键的Ru@TiO2催化剂。界面金属键有效地促进了金属氧化物与金属间的电子转移,促使Ru簇的价带中充满电子,使其具有最优HBE的同时,难以被氧化且CO吸附受阻,具有很强的抗CO中毒特性和抗氧化性。该催化剂在酸性和碱性条件下,HOR质量活性比商业化的PtRu/C催化剂高15%至30%。
此外,催化剂表面含氧物种的吸附也会影响表面H的吸附强度,从而改变HOR反应机理。Feng等65通过DFT理论计算发现,Pt(110)表面吸附的OH*会通过电子效应调节中间物种的吸附(图7a,b)。主要表现为增强相邻金属位点H2O*的吸附,而削弱H*的吸附。OH*覆盖度越高,其对H2O*吸附的增强效应和H*吸附的弱化效应就越显著。OH*对H2O*的吸附增强效应可归因于两个方面,一是源于OH*引起的H2O*与Pt(110)表面的内在相互作用的增强;另一方面是来自于OH*与H2O*之间的氢键作用,氢键的键能大约是0.187-0.218 eV。据此,他们认为H*的吸附自由能(Δ GH*)并不是唯一的HOR/HER活性描述符。OH*的形成电位(UOH*)不仅可以表征催化剂的亲氧性,还可用于区分HOR/HER机理的变化和活性描述符的适用范围。如图7c,d所示,对Pt(110)或PtRu(110)而言,当电势低于UOH*时,催化剂表面难以形成OH*,ΔGH*可作为唯一的HOR/HER活性描述符;当电势高于UOH*时,催化剂表面吸附的OH*抑制了H2O*的脱附,此时水的脱附决定了HOR活性,即水的吸附自由能(ΔGH2O*)成为活性描述符。该计算也证实,PtRu(110)催化剂中Ru和Ru-OH*均可以调节邻近Pt位点的电子结构,使其对H的吸附能达到最优值,且抑制Pt上OH*的形成。

图7 不同金属表面有无OH*时(a) H*和(b) OH*的吸附能;(c) Pt(110)和(d) PtRu(110)上不同电极电势(U,V vs NHE)下,有无OH*时,HOR基本反应步骤和OH*形成的吉布斯自由能变图Fig. 7 Adsorption energies of (a) H* and (b) H2O* on different metals surfaces with and without OH*;Gibbs free energy change of HOR elementary reaction steps and OH* formation at varied electrode potentials (U, V vs NHE) on (c) Pt(110) and (d) PtRu(110) with and without OH*.
3 电化学界面的理论模拟
为了深入研究HOR/HER的电化学过程,从原子、分子尺度探究在电场作用下电极/电解质界面上的电荷转移和成键断键的过程,需要对电极及界面区域的液体电解质(包括电化学双电层的紧密层和分散层)进行更加透彻的分析。原位界面结构表征和第一性原理的理论计算都可以在一定程度上明确电化学界面的结构和性质。实验表征HOR/HER反应机理因易受pH、离子、温度等外部环境和实验条件等因素的影响,其宏观的反应机理和催化行为往往是多种因素共同的作用产生,为探究影响HOR/HER活性的本质原因提出了挑战。理论计算的发展不仅可以为电化学界面给出相应的模型,还可以模拟电化学系统,为复杂的界面过程提供分子尺度的信息,弥补了实验的不可控性和复杂性。该部分将从理论计算的角度出发,初步介绍目前界面电化学的理论模拟方法及其在HOR/HER中的应用。
电极界面双电层(the electric double layer,EDL)结构的第一个模型建立于一个多世纪以前,原子尺度上界面结构的详细数据直到20世纪80年代至90年代才出现,且当时只是将超高真空表面科学技术应用到电化学领域65-70,无法将动态溶剂化效应、pH效应、阳离子效应和界面电极电势等环境因素对电化学过程的影响考虑在内。随着理论和技术的不断发展,理论计算和模拟对电化学界面和电化学反应的描述越来越完善,近年来才发展出对EDL更加精确的模拟71-76。由于电极表面涉及到成键断键、电子转移以及溶剂的动态变化等复杂过程,简单的静态结构计算无法完全考虑所有过程,动力学的计算方法成为理想选择。经典的分子动力学(molecular dynamics,MD)可以在较长时间和空间尺度内计算体系内的色散相互作用77,78,确定界面水分子与金属材料的相互作用79-82,但不能模拟固体表面的反应性。从头算分子动力学(ab initio molecular dynamics,AIMD)虽然无法模拟较长时间和空间尺度的反应,但可以在每一个时间步长内执行一个完整的电子结构量化计算,所得的计算结果的精度比MD高很多。DFT计算表明金属电极附近的水分子存在强极化,电荷会从水向金属转移75,83,这意味着准确的电极/电解质界面建模不仅需要通过分子动力学模拟来考虑电解质的液体性质,还需要正确的电子结构计算来确定界面电子的极化效应。AIMD可对体系进行动力学分析的同时,通过第一性原理计算确定每一个时间步长内的几何和电子结构,成为一种模拟电化学界面的有效方法。
探究电极表面双电层中电极与电解质之间的相互作用是了解电极界面结构的关键。电极界面模型的建立主要面临两个挑战:(1)合理地考虑电解质的性质;(2)界面电极电势的控制。针对第一个挑战,有研究者采用将电极与半经验的隐性溶剂化模型结合的方式来模拟界面,但该数值的方法来表示液体电解质有一定的局限性84,尤其是当电解质溶液中发生明显的电荷重组时85。因此,电极/电解质界面的模拟需要考虑显性溶剂化模型。最初,研究者在考虑金属电极和含水电解质之间的界面结构时假设水为冰状结构86-88。后期,人们在模拟了液态水层的界面后83,得到了与实验测量值更加接近的计算结果。例如,Groß等75对包含有144个水分子的H2O/Pt(111)界面进行了AIMD模拟(图8a),探究了有、无水层时原子的运动轨迹以及表面功函和静电势随时间的变化(图8b),计算所得的电极零电荷电势与实验值吻合(分别为4.96和4.9 V)。如图8c所示,他们还发现水膜的存在会导致静电势的变化,靠近电极的静电势不仅受水分子层结构控制还受水分子极化的影响,部分电荷会从第一水层向Pt电极转移。这表明正确描述双电层确实需要量子化学的处理方法,也表明双电层应视为一个由界面处原子结构的电荷分布所决定的内部电场89。Szabová等90则利用AIMD揭示了溶剂化对Pt6/CeO2/H2O体系的催化反应活性和电子性能的影响,为真实条件下的表面电催化反应的理论计算提供了新的思路。Bellarosa等91采用第一性原理BOMD (Born-Oppenheimer molecular dynamics)计算发现催化剂表面水分子的反应性与金属相接区域的结构密切相关,通过径向分布函数、界面形态和相关结构分析发现不同的贵金属的水/金属界面具有不同的反应性。也有研究者将显性和隐性溶剂化模型结合92,93,但涉及到隐性溶剂化模型时,计算结果的正确性还需要更加可靠的参考文献来验证。因此,对于电极/电解质界面结构的性质,理想的模拟方法是对电化学界面的结构和过程进行大规模的显性溶剂化AIMD模拟,但此类模拟所耗成本较高。

图8 零电荷电势平衡过程中的(a)快照和(b)功函随时间的变化;(c)真空下Pt(111)的静电势V(z) (虚线)和带无离子水膜的Pt(111)电极的平均电势(实线),下图表示了由水膜造成的变化Fig. 8 (a) A snapshot and (b) time evolution of the work function along the trajectory to determine the potential of zero charge equilibrated; (c) electrostatic potential V(z) of Pt(111) in a vacuum (dashed line) and the averaged potential of the Pt(111) electrode with an ion-free water film (solid line). The charge caused by the water film δV(z) is illustrated in the lower panel.
针对第二个挑战——界面电极电势的模拟,Lozovoi等94提出了一种通过引入补偿电荷来实现电中性的恒电势计算方法,但该方法可能会出现因为电子数量在自洽场循环的波动引发收敛性问题。Bonnet等95也提出了一种方法,将电子与恒电势器的虚拟交互进行交换,使得电极电势在分子动力学运行过程中围绕着一个期望值震荡,而在电子结构计算中,每一步的电荷都保持恒定。近期,该方法在一定程度上得到了发展96,97,但是仍然存在一个关键性的问题,即如何选择一个适当的参考电势值95-98。Cheng等53采用量子力学分子动力学(quantum mechanics molecular dynamics,QMMD)进行了H2O/Pt(100)界面的模拟(图9),通过在显性溶剂中加入Na+(图9b),以及在显性溶剂的上方加入隐性溶剂(图9c)等方案99向体系引入额外电子,即通过在Poisson-Boltzmann近似下的显性/隐性溶剂界面处引入分数电荷来控制功函,从而实现电势的变化。例如,通过引入0.97、1.94、3.06和3.48 e-的额外电子,使得功函值分别为4.73、4.34、4.11和3.98 eV,从而模拟电势为+0.29、-0.10、-0.33和-0.46 V的电极表面。当H2O/Pt(100)的功函为5.06 eV时,对应的U值为0.62 V,接近于实验条件下Pt(100)的零电荷电势值0.41 V。

图9 QMMD模拟中的H2O/Pt(100)的界面模型:(a)显性溶剂模型(~6层水);(b)显性溶剂+ Na+模型(~层水+ 1Na);(c)显性+隐性溶剂化模型(~3层水+隐性溶剂)Fig. 9 Models of water/Pt(100) interface in QMMD simulation. (a) Explicit model (~6 water layers), (b) explicit + Na+model (~6 water layers + 1Na) and (c) explicit + implicit model (~3 water layers + implicit solvation).
对碱性介质中HOR/HER反应机理的理论探究,目前大部分仍主要采用基于DFT的热力学计算,着眼于研究HOR/HER基元反应的自由能变。例如,Norskov等67利用DFT计算了大量过渡金属表面的HOR和HER的三个基元步骤Tafel、Heyrovsky和Volmer的吉布斯自由能变。结果表明,Pt(111)电极上的HOR/HER的决速步是Tafel步骤,当U = 0 V (vs NHE)时,反应能垒为0.85 eV,与实验值0.2 eV有一定差异,但是根据该反应能垒计算得出的交换电流密度与实验值完全一致。他们还发现H吸附自由能是描述电极表面HOR/HER活性的最重要参数,用H吸附自由能拟合的交换电流密度遵循火山关系曲线,且与实验数据吻合良好。
关于碱性介质中HOR/HER反应动力学的研究也是科研工作者关注的焦点,通过微观动力学模拟,可对影响反应机理及动力学的关键因素进行定量描述。Strmcnik等35通过微观动力学模拟Au、Pt和Ir电极的HOR/HER极化曲线,并研究具有pH依赖性(H3O+、OH-)和不具有pH依赖性(H2、H2O)的反应物浓度对极化曲线的影响。研究发现当pH为9时,Pt和Ir的HOR极化曲线中存在两个明显的传质控制平台,表明总电流受两个独立过程(物种)的影响;当pH从11到9.5时,随着OH-浓度的减小,Pt和Ir的极限扩散电流密度减小,证明OH-在HOR中起着重要作用,但具体地OH-如何影响HOR反应动力学,其微观层面影响机制仍不明确。
DFT计算与微观动力学的结合是深入研究HOR/HER机理的发展方向。有研究者将DFT计算结果作为微观动力学模型的基本参数,对HOR/HER反应机理及动力学有了更深刻的认识。例如,Huang等12基于DFT计算的反应自由能变、H+的电荷、OH-的溶剂配位数等参数,建立了碱性Volmer步的微观Hamiltonian模型,其中包含了电子相互作用、键的断裂、溶剂重组和双电层效应。该模型给出了一个简单但是信息量大的碱性Volmer步的活化能垒公式,量化了各因素的贡献,并阐明了在带电量更大的界面上活化能更高,不是因为溶剂重组更难,而是OH-达到双电层区域需要做更大的功。Lamoureux等100则基于DFT理论,建立了具有简单扩散控制的HOR/HER微观动力学模型,计算所得的HOR/HER理论活性与实验所得的极化曲线和Tafel斜率在定性上是一致的,且认为HOR/HER活性随pH值的变化是质子供体发生变化所引起的。
显然,理想的电极/电解质界面HOR/HER反应机理与反应动力学的计算,需要建立合理的显性溶剂化模型,通过AIMD与DFT计算表面性质,并结合微观动力学模型来分析催化剂表面的HOR/HER动力学参数的方式来实现。该方法可以在充分考虑液体电解质中存在的动态溶剂化效应、pH效应、阳离子效应的前提下,通过电子结构计算来确定电极/电解质界面电子的相互作用,对界面静态结构、反应过程中双电层中反应物种与溶剂等的动态结构进行深入研究和分析,最终得到催化剂表面性质、活性以及反应机理的微观层面的全面描述。
4 结论与展望
碱性介质中的HOR/HER反应速率较酸性介质中慢2至3个数量级,极大地限制了碱性燃料电池与电解水的发展。深入研究碱性介质中HOR/HER反应机理,明确酸、碱性反应动力学差异之根本,是开发低成本、高活性和高稳定性HOR/HER催化剂的关键。
本文对近年来碱性介质中HOR/HER的相关机理研究进行了总结,其相应反应机理的解释主要包括:HOR/HER的双功能机理、HBE理论和电子效应。双功能机理强调了水的解离以及OHad分别对HER和HOR的影响,对设计制备复合催化剂具有指导意义。HBE理论则强调Had是HOR/HER的关键,其他外部因素,如电势、溶剂等的改变对HOR/HER的影响都是通过对HBE的调节得以实现。HBE是HOR/HER唯一的活性描述符,也为催化剂活性的调节提供了便捷的指示参数,但需进一步研究各外部因素对HBE影响的机制和本质原因。电子效应则强调催化剂表界面组成、反应中间物种等对活性位电子结构的影响,对构建最优活性位提供了优化策略,但活性位电子结构的调节依赖于HOR/HER反应机理。由于电催化反应体系的复杂性,以及电极/电解质界面上催化剂表界面的电子结构、双电层结构对电极电势和溶剂的依赖性,导致对HOR/HER反应机理难以采用单一理论进行合理的解释,并对研究影响HOR/HER活性的本质原因提出了极大的挑战。
理论计算的发展可为复杂的电催化反应界面过程提供原子、分子层面的信息,可以弥补实验的不可控性和复杂性。深入的HOR/HER反应机理的理论模拟,需要在理想的电极/电解质界面建立更加合理的显性溶剂化模型,通过AIMD与DFT等方法相结合计算电极表面电子结构以及双电层中的反应过程中键的断裂与形成、溶剂的重组、质子的迁移等动态结构,并结合微观动力学模型来分析不同电极电势下催化剂表界面的HOR/HER机理与反应动力学。综合现有的研究可以发现,实验与理论计算的结合,原位界面结构表征和理论模型的发展,为理清碱性介质中HOR/HER机理、研究pH效应的本质原因提供了可能,有望为设计制备低成本、高活性、高稳定性HOR/HER催化剂提供应对策略和理论依据。
猜你喜欢
建材发展导向(2021年14期)2021-08-23
防爆电机(2020年4期)2020-12-14
河北理科教学研究(2020年1期)2020-07-24
中国煤层气(2019年2期)2019-08-27
中国有色金属学报(2018年2期)2018-03-26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
环境与可持续发展(2017年2期)2017-04-06
浙江农业科学(2016年11期)2016-05-04
文物保护与考古科学(2016年1期)2016-04-16
高中生学习·高二版(2014年5期)2014-07-03

- 物理化学学报的其它文章
- 有序金属间化合物电催化剂在燃料电池中的应用进展
- Recent Progress in Proton-Exchange Membrane Fuel Cells Based on Metal-Nitrogen-Carbon Catalysts
- 提升燃料电池铂基催化剂稳定性的原理、策略与方法
- 高温聚合物电解质膜燃料电池膜电极中磷酸分布及调控策略研究进展
- Formic Acid Electro-Oxidation Catalyzed by PdNi/Graphene Aerogel
- Enhanced Performance and Durability of High-Temperature Polymer Electrolyte Membrane Fuel Cell by Incorporating Covalent Organic Framework into Catalyst Layer