
锂离子电池正极材料合成及改性
2021-09-28 03:36王策王国庆王二锐吴天昊尉海军
化工进展 2021年9期
王策,王国庆,王二锐,吴天昊,尉海军
(1 北京工业大学材料与制造学部先进电池材料与器件研究所,北京 100124;2 新型功能材料教育部重点实验室,北京 100124)
化石能源的大量消耗加剧了能源短缺与环境危机,使人类生存环境面临威胁,因此急需开发新型环保且可持续的能源。其中化学电源是一种能量转换效率高、环境友好的储能装置,逐渐进入人们的日常生活。作为化学电源一员的锂离子电池不仅具有能量密度高、环境友好和循环寿命长等优点,而且已经在3C 电子产品、电动工具、电动汽车和其他交通工具等方面广泛应用[1-3]。国家对新能源汽车的政策推动,必然会带动锂离子电池及其相关产业的快速发展。
新能源汽车行业的快速发展以及对电动汽车长续航里程的不断追求,迫使人们对锂离子电池的能量密度提出了更高的要求。这就要求锂离子电池正负极材料容量足够高,与负极材料相比,正极材料的容量相对较低,因此核心问题是提高正极材料的容量。目前商业化的正极材料主要有钴酸锂(LiCoO2)、三元材料镍钴锰酸锂(LiNi1-x-yCoxMnyO2,NCM)/镍钴铝酸锂(LiNi1-x-yCoxAlyO2,NCA)、锰酸锂(LiMn2O4)和磷酸铁锂(LiFePO4)等,这些材料的容量普遍低于250mA·h/g,远低于负极材料的容量。高镍层状氧化物正极材料(NCM/NCA,Ni≥80%)经过数年的不断发展,在市场上开始普及应用,但Ni 含量的增加也降低了锂离子电池循环寿命及安全性[4]。高容量(>250mA·h/g)和低成本的富锂锰基层状氧化物正极材料[LLOs,xLi2MnO3·(1-x)LiMO2]成为下一代高容量正极材料的候选材料之一。但是一些固有的问题阻碍其商业化应用,如首次库仑效率低、倍率性能差、循环性能差及严重的电压衰减等问题,需进一步研究[5-10]。除掺杂和表/界面处理等常规改性措施外,研究人员在球形团聚体材料制备工艺的基础上开发了单晶和浓度梯度材料来改善正极材料的一些固有问题。本文以前体合成为出发点,分别对球形团聚体、单晶和浓度梯度三种正极材料进行了详细的研究,综合各种材料的优缺点,为正极材料的发展提供了一些参考。
1 前体合成
前体的合成方法主要包括水热/溶剂热法[11]、溶胶-凝胶法[12]、喷雾热解法[13]和共沉淀法[14]等。其中共沉淀法由于工艺简单、元素分布均匀、形貌和粒径易调控且可大规模工业化生产,故高容量正极材料多采用此方法合成。锂盐的溶度积相对过渡金属盐较大,很难实现与过渡金属离子一起沉淀,一般优先制备过渡金属前体,然后进行锂化烧结得到最终的正极材料。故获得较好的前体是制备性能优异的正极材料的关键。
反应开始前在恒温的反应釜中预先添加一定量的底液,为沉淀反应提供一个合适的化学环境。反应开始时,将过渡金属盐溶液、沉淀剂和络合剂共流到反应釜中,在一定的搅拌速度下,经过颗粒成核、生长、团聚且相互碰撞、抛光等物理化学过程,生成内部致密、表面光滑的球形颗粒[15]。反应过程中持续通入惰性气体来抑制金属离子的氧化。反应结束后经过一定时间陈化,浆料过滤、洗涤和烘干,最终得到前体。
1.1 反应机制
共沉淀反应是晶核形成(成核)和晶体生长共同作用的结果,反应始于成核,成核伴随着生长。Dahn 和Yoon 课题组[16-17]系统研究了共沉淀反应机制,结果表明,反应初始阶段金属离子在溶液中优先与氨络合,再慢慢地释放,与沉淀剂进行反应,反应体系达到络合与沉淀的动态平衡,整个反应过程中伴随着一次粒子团聚、吸附和溶解-再结晶、二次颗粒不断长大。如图1所示,反应主要包括成核和生长两部分[18]。

图1 前体反应机制示意图
1.1.1 成核
成核涉及许多复杂的物化反应,且Ni2+、Mn2+和Co2+对应沉淀盐在不同pH环境下的溶度积不同,为保证合成的前体沉淀反应完全、元素分布均匀且无富集,常常加入氨水为络合剂,将不同的过渡金属离子络合,降低过饱和度,从而降低成核速率,达到三种过渡金属离子共同沉积的效果。络合和沉淀反应如式(1)、式(2)所示[18]。

反应初期,过渡金属离子优先与氨水形成多种配位的络合物,然后络合物缓慢释放金属离子,并与沉淀剂进行沉淀反应成核。络合反应与沉淀反应逐渐达到动态平衡,生成大量的晶核,整个反应体系中氨水的浓度和pH 对成核起着决定性的作用。Cheng 等[18]通过搭建模型与理论计算研究了氨水的浓度和pH对Ni2+、Co2+和Mn2+反应过程中存在形式及浓度的影响,研究结果表明,随氨水浓度和pH增加,Co2+和Mn2+易完全沉淀,而残留的多种形式的Ni2+络合物较多。高镍正极前体Ni含量较高,在前体合成过程中应严格控制氨水浓度和pH,减少溶液中残留的含Ni2+络合物,使过渡金属盐溶液完全沉淀。对于富锂锰基层状氧化物材料,由于Mn(OH)2容易被氧化成MnO(OH),所以常常以碳酸盐作为沉淀剂进行反应[19]。Belharouak 等[20]研究Ni0.3Mn0.7CO3的生长机制发现,反应初期,颗粒粒径、化学组分和结构形貌均不均匀,反应始于形成碳酸盐主相和羟基氧化镍杂相的混合物。反应一段时间后,形成化学组分均匀,且具有良好的结构和形貌的颗粒。此外,根据成核速率公式[式(3)][18]。

式中,k为常数;NA为阿伏伽德罗常数;M为生成物摩尔质量;σ为固-液界面张力;ρ为生成物密度;S/S0为过饱和度;T为绝对温度;R为理想气体常数。
由式(3)可知,成核速率与温度及过饱和度成正比,与固-液界面张力成反比。过饱和度越大,成核速率越快,不利于晶体生长,反应易生成大量无定形絮状物,pH 和氨水浓度增加会导致过饱和度增大,从而产生许多细小晶粒。另外,由于成核和生长相互作用,当成核速率较大时,会抑制晶粒长大,形成许多细小晶粒,随着体系反应过程不断进行,前期产生的大量亚稳晶粒才会慢慢生长成晶体[21]。
1.1.2 生长
随着反应进行,一次颗粒逐渐生长,在搅拌的作用下相互之间的碰撞概率增加,且团聚能够有效降低表面能从而有助于体系能量的降低[22]。因此,在范德华力(与颗粒尺寸有关)与湍流聚集(与颗粒表面有关)的作用下,一次颗粒之间互相吸引团簇,形成二次颗粒。络合作用导致部分晶粒溶解,沉淀作用助于重新结晶,体系达到络合-沉淀的动态平衡。一次颗粒生长、二次颗粒吸附一次颗粒及二次颗粒之间相互团聚使得颗粒粒径不断长大[15]。一次颗粒与二次颗粒的粒径和形貌及一次颗粒的团聚方式决定了前体的形貌结构、比表面积和振实密度等物化性能,从而直接影响正极材料的电化学性能。
无论是成核还是晶体生长,都与反应体系中氨水浓度和pH有关,氨水浓度和pH仍是晶体生长的主导性因素,pH 增大,沉淀速度大于络合速度导致快速形成许多晶粒,团聚和吸附变困难,二次颗粒难以长大导致粒径减小。氨水浓度增大,络合增强,成核速率降低,生长变慢,粒径也会减小。
1.2 合成工艺
共沉淀工艺受诸多因素影响,pH、氨水浓度、合成温度和反应时间等主要工艺参数对前体粒径及粒径分布、形貌、结晶程度、比表面积和振实密度产生影响。均一组分球形前体和梯度前体主要在于组分的不同,故前体的合成也有所差别。各种影响因素对不同类型前体的影响在下文逐一进行阐述。
1.2.1 均一组分球形前体
均一组分球形前体的合成工艺是共沉淀工艺中合成最早也是最成熟的技术,目前工业中普遍应用的沉淀剂主要包括Na2CO3(Na2CO3溶液)和NaOH(NaOH 溶液)等。通过研究pH对两种沉淀体系中的游离过渡金属离子浓度和沉淀转化的吉布斯自由能(ΔG)的影响发现[23-24],碳酸盐体系pH 在7.5~8.5 之间三种过渡金属离子能够共同沉淀完全,氢氧化物体系pH在10.5~11.5时三种离子沉淀转化最容易进行,如图2。Sun 等[14]研究发现在一定范围随着pH 增加,颗粒粒径减小但压实密度增加。同时适当地提高温度有利于提高反应活性,Zhang等[25]研究了温度对前体[Ni1/3Co1/3Mn1/3]CO3的影响,发现随着温度升高,XRD 峰强增加、峰形尖锐且峰宽减小,表明提高温度有利于晶体生长,减少体系中细小颗粒。但70℃时观察到Mn3O4或MnO2杂相,因为高温导致氨等物质挥发,氧化了前体中的Mn2+,如图3。除此之外,Zhang 等[25]和Sun 等[14]系统地研究了氨水浓度对前体形貌及结构的影响,发现氨水浓度增大有助于球形度和结晶度增加,但浓度过高会出现裂纹和杂相,且氨与Ni2+的强配位生成的[Ni(NH3)n]2+会引发Ni2+沉淀不完全等问题,如图4。Yu 等[26]通过改变氨水/过渡金属盐溶液比例,增大了前体的振实密度,从而提高了正极材料循环性能并缓解了循环过程的电压降。

图2 pH对球形前体合成的影响

图3 合成温度对球形前体合成的影响

图4 氨水浓度对球形前体合成的影响
综上所述,球形前体的合成过程中,pH、氨水浓度、合成温度和反应时间等因素之间相互影响,反应过程体系处于络合-沉淀的动态平衡之中,且不同组分的前体所需最佳工艺参数均有所不同。此外,氨水还会对环境造成污染,所以对前体合成工艺的优化十分重要。
1.2.2 单晶材料前体
单晶材料前体与球形多晶材料前体的合成工艺流程相似,但工艺参数略有不同。首先,单晶材料可以看作是一种结晶度高、微米级或亚微米级尺寸的一次颗粒,且具有某些晶面的优先暴露和单一分散的特性。为了制备性能优异的单晶材料,前体的尺寸不宜过大,Sun 等[27]通过共沉淀法合成了适合制备单晶材料的前体,该前体的一次颗粒大小为10nm,并团聚为尺寸2µm的二次颗粒。由此可见,该前体与上面所述的球形前体形貌完全不同,参考pH、氨水浓度和反应时间对前体的影响,所以单晶材料所需前体的合成一般需要较高的pH、较低的氨水浓度和较短的反应时间等条件[28]。
1.2.3 浓度梯度前体
浓度梯度前体的合成工艺与均一组分前体的合成工艺不同之处主要在于盐溶液的控制,某些元素浓度会随着反应而产生浓度梯度变化,所以需要额外的反应泵将特定比例的外层壳的过渡金属盐溶液加入到内部核的过渡金属溶液,均匀混合后再泵入到反应釜进行共沉淀反应,如图5所示。根据元素浓度斜率变化可将浓度梯度分为半浓度梯度[内层(核)或外层(壳)为梯度]、单斜率全浓度梯度和多斜率全浓度梯度三种[29-31]。合成半浓度梯度材料时,先将盐溶液1泵入到反应釜中沉淀反应一定时间后,再将特定比例的盐溶液2泵入到盐溶液1中混合均匀再共流到反应釜中进行反应,形成具有梯度壳的前体;或先将特定比例的盐溶液2泵入到盐溶液1中的同时再共流到反应釜中进行反应,待盐溶液2全部泵入盐溶液1中后,再持续将混合溶液泵入到反应釜中持续反应一定时间,形成具有梯度核的前体。单斜率全浓度梯度前体合成时,在反应开始时就将特定比例的盐溶液2 泵入到盐溶液1中,混合均匀的同时泵入反应釜中进行反应,一直持续到反应结束,形成从核到壳的全浓度梯度前体;多斜率浓度梯度合成时,需要盐溶液2泵入到盐溶液1中的同时再共流到反应釜中进行反应,待盐溶液2完全泵入到盐溶液1中之后,再将盐溶液3泵入盐溶液1中,继续共流到反应釜中持续反应,形成多梯度斜率的前体。

图5 浓度梯度前体合成
浓度梯度材料具有较高容量同时兼顾良好的结构稳定性,其元素的浓度梯度分布源于前体合成过程的设计,所以浓度梯度前体的合成工艺至关重要。合成浓度梯度前体时不仅要通过微积分公式精确计算梯度溶液组分,同时随着某些元素浓度的变化,还要精准调节沉淀剂和络合剂,维持体系的动态平衡。
1.3 前体晶面取向
研究发现不同晶面上的Li+扩散速率差异较大,常见的层状氧化物正极材料的{010}晶面上的Li+扩散较快,而{001}晶面Li+传输速度较慢[32-33]。若能调控一次颗粒晶面择优取向,使活性晶面大面积暴露,可显著提高材料的电化学性能。Wu 等[34]通过调整共沉淀工艺,合成了特定晶面暴露的前体,而后预氧化前体,锂化烧结后材料{010}中活性晶面被保留,5C放电容量在140mA·h/g 以上,且循环性能优异[图6(a)~(d)]。在前体的制备过程中引入表面活性剂能够影响不同晶面的生长速率,从而调节特殊晶面生长[35],如图6(e)。Sun 等[36]利用NaOH 为沉淀剂、氨水为络合剂且在共沉淀过程中加入表面活性剂PVP 来调控前体的晶面取向,利用表面活性剂PVP 在不同晶面上的吸附速率不同从而影响晶面的生长速率的特性,最终得到{010}活性晶面暴露的六方纳米砖型前体Ni1/3Co1/3Mn1/3(OH)2,锂化烧结后材料的倍率和循环性能较好。通过调整共沉淀工艺,或在共沉淀工艺过程中加入吸附剂可以有效地促进某些活性晶面的生长。

图6 {010}中活性晶面暴露的Li1.2Ni0.2Mn0.6O2表征及NCM111纳米砖制备示意图
晶面取向调控与沉淀过程中的成核和生长有关,进而与体系的能量有关。某些晶面的表面能较高导致晶面不稳定,通过晶面生长可以有效降低能量、稳定晶体;某些晶面的吸附能较高,导致晶面上沉积更多的离子,促进晶面生长,从而降低能量[32]。
2 正极材料制备
工业上一般将前体与锂源混合均匀,采用高温固相法制备正极材料,主要影响因素包括锂源和烧结工艺等。球形团聚体、单晶和浓度梯度材料的制备又有各自的特点。
2.1 球形团聚体
Li含量和烧结温度不仅影响颗粒形貌,还对材料的结晶程度、晶格排列和相结构比例有重要影响。Liu 等[37]研究了球形团聚体Li1+xNi1/6Co1/6Mn4/6O2.25+x/2系列材料,如图7(a)随着Li含量的增加不仅破坏了前体的球形结构,而且增大了一次颗粒粒径,当x=0.3 时材料的电化学性能最佳。此外,Mai 等[38]对Li1.098Mn0.533Ni0.113Co0.138O2研究发现,富锂锰基材料在制备过程中适当降低Li含量使材料形成氧空位,能够提高Li+扩散动力学,并且在表面形成尖晶石相从而提高电化学性能。Kang等[39]利用退火工艺调整0.5LiNi0.5Mn1.5O4·0.5Li2MnO3材料内部结构比例,材料经退火后形成了含缺陷的尖晶石相和阳离子无序的层状相,含缺陷的尖晶石相可以缓解结构畸变,而无序层状相提高了电化学活性,从而提高了放电容量和倍率性能,首次放电容量大于300mA·h/g,10C放电容量为160mA·h/g。

图7 球形团聚体与单晶材料的SEM图
高镍NCM 材料制备时由于烧结过程Li 挥发,故一般Li过量。Ni2+与Li+离子半径相近,烧结过程中易发生离子混排,故烧结过程中常常通入O2来抑制过多的Ni2+形成。Pan 等[40]认为适当的提高Li/TM能够增加Li+/Ni2+混排并改变Li 层间距从而提高动力学来改善电化学性能。
综上所述,球形团聚体材料的制备应根据不同组分确定最佳的Li 含量和烧结工艺,精准调控材料内部的相结构比例和晶格占位,提高材料物化性能和电化学性能。
2.2 单晶材料
与球形团聚体相比,单晶材料缺陷相对较少、比表面积小、机械强度大,能够有效降低副反应。常见的制备方法主要有高温固相法和熔盐法等[36]。高温固相法是指直接将前体与锂源混合而后在高温度下煅烧,经破碎直接得到单晶材料的方法,Dahn等[41]利用高温固相法制备了颗粒粒径为2~5µm的单晶LiNi0.8Mn0.1Co0.1O2材料[如图7(b)],该材料具有较高的放电容量和优异的循环性能,首次放电容量为240mA·h/g,且循环2000 多次后保持率大于90%。熔盐法是在烧结过程中利用熔盐(NaCl、KCl 和CsCl 等)制备单晶材料的方法,Yu 等[42]以KCl为熔盐合成了单晶富锂材料[如图7(c)],并通过表面修饰改善了电压衰减,提高了材料的热稳定性和安全性。高温固相法制备单晶材料工艺简单,可大规模工业化生产,但高温会引起过量锂挥发,且过多的锂可能导致严重的团聚。熔盐法能够降低烧结温度、减少阳离子混排和颗粒团聚,且通过熔盐的选择可以调变晶面取向来改善电化学性能,但产量较低且需要水洗去除多余的盐导致较高的成本[43]。
单晶材料机械强度高,能够增大极片压实密度提高能量密度,同时缺陷和比表面积相对较小,能够降低副反应、抑制颗粒粉化,提高材料的长循环稳定性。
2.3 浓度梯度材料
浓度梯度材料由于其从内到外逐渐变化的元素分布,在烧结工艺上相较于均一组分的材料也应略做调整和探索。对于梯度材料来讲,存在最明显的现象就是高温烧结过程中造成的元素扩散[图8(a)和图8(b)]导致组分的偏移[29],这在众多梯度相关的工作中均有体现。另外,Chen 等[44]通过高温原位XRD 研究了浓度梯度LiNi0.6Mn0.2Co0.2O2材料的烧结过程,并对烧结工艺进行探索,他们认为预烧过程和烧结温度对于控制相结构分离和减小表面Li2CO3组分至关重要。其次,Yu 等[45]制备了全浓度梯度斜率依次增大的三种富锂锰基材料S1、S2和S3[如图8(f)],分析三种材料的高温原位XRD可知,随着浓度梯度斜率增大,层状结构向尖晶石结构转变的温度升高,分别为245℃、285℃和325℃,表明梯度材料有效增强了结构稳定性。但是浓度梯度斜率过大时材料中的电化学应力和本征应力会负作用于电化学性能。

图8 前体和锂化烧结材料从颗粒中心到表面的过渡金属原子的分布及三种不同的单斜率浓度梯度材料的元素浓度变化和高温原位XRD
浓度梯度材料的制备过程中,首先应注意减缓高温烧结造成的元素扩散以保证元素的梯度分布,另外要抑制Li+/Ni2+混排、降低表面Li2CO3,同时还需减缓烧结之后存在于材料中的应力并且避免相分离。所以应严格控制烧结温度、升温速率和烧结时间等。全浓度梯度材料具有较高容量同时兼顾良好的结构稳定性,且随着电化学循环有效地缓解结构破坏。制备浓度梯度材料时应综合物化性能和电化学性能,既要保持较高的容量和循环稳定性,同时也应考虑材料的应力和热稳定性。
高容量正极材料应用从球形团聚体开始,伴随着循环过程中性能衰退等一系列问题,人们开始研究结构更加稳定的单晶和浓度梯度材料,单晶材料比表面积和结构缺陷小、机械强度大,能够减小副反应和颗粒破碎从而提高了长循环稳定性,但是也存在容量不高和倍率性能不佳的缺点。浓度梯度材料将某些元素进行梯度分布,设计初衷是为了保证材料的高容量和长循环稳定性。总的来说,单晶和浓度梯度材料表现出更好的长循环稳定性,能够有力地推动正极材料的发展。
3 正极材料改性
正极材料能量密度不断提高,使得稳定性和安全性等问题更加突出,掺杂和表/界面改性能够有效缓解这些问题。
3.1 元素掺杂改性
元素掺杂主要是将异质元素引入晶体结构中占据锂、过渡金属或氧的位置,可以增强材料的导电性和结构稳定性,进而提高倍率性能和缓解电化学循环过程中的结构转变。根据掺杂形式分为体相掺杂、表面掺杂和梯度掺杂。
3.1.1 体相掺杂
过去几十年中元素掺杂技术研究较多,成为材料改性的有效手段之一,并且主要集中于体相掺杂的研究。适当浓度的离子掺杂可以有效缓解材料体相结构的不可逆转变,进而提高材料的稳定性。Xin等[46]计算了待掺杂元素与基体元素Ni、Co、Mn的相容性及与O 形成TMO 的结合能,结果表明Sc、Ti、V、Cr、Fe、Y、Zr、Nb 元素掺杂可能有效。Wang等[47]以Li2Mn0.75M0.25O3为模型,通过DFT计算研究了M的电子结构、O2释放吉布斯自由能、M-O结合能与M的电荷转移倾向,发现Nb为最佳的掺杂元素。Yu 等[48]在LLOs 材料中的Li+位引入Nb5+,结合理论计算发现Nb5+掺杂进入Li位能够降低Li+的迁移能、稳定结构,提高电化学性能。离子掺杂还可以宏观调控一次颗粒形貌,来提高材料结构的稳定性。Sun等[49-50]使用B和Ta等元素掺杂到高镍材料中,通过调控一次颗粒得到放射状的二次球形团聚体,该特征材料缓解了晶格变化产生的应力集中,进而维持结构的稳定性。同时,他们还研究了F-引入到梯度三元材料Li[Ni0.80Co0.05Mn0.15]O2中得到阳离子有序的富镍层状材料,有效地抑制深电荷状态下的微裂纹的形成与扩展,使用软包电池在25℃下循环8000次后容量保持率为78%,表现出优异的循环稳定性[51]。异质元素引入体相结构,在电化学循环过程中可以有效地支撑结构,缓解不可逆相转变和维持结构稳定性,提高材料的电化学性能,但是过量的非活性元素掺杂会明显降低材料的容量。
3.1.2 表面掺杂
对于高容量正极材料,尤其高压状态时材料表面结构的稳定性尤为重要。相对体相掺杂来说,表面掺杂的作用在于表面建立保护层保护材料表面的结构稳定性。Wang 等[52]将Li1.2Mn0.54Ni0.13Co0.13O2分散于含乙醇铌的乙醇溶液中,超声、干燥后于600℃中热处理,实现了表面几纳米厚度的Li位Nb掺杂,如图9(a)。在表面形成了稳定的类岩盐相,引入的Nb-O 抑制表面氧的活性,提高了循环稳定性,循环100 周容量保持率为94.5%,远高于未掺杂样品的76%,电压降仅为136mV。另外掺杂元素不仅能取代其他原子位置,还能占据空位。Guo 等[53]利用NaCl 熔盐法对0.5Li2MnO3·0.5LiNi1/3Co1/3Mn1/3O2进行Na+表面梯度掺杂[如图9(b)],大离子半径的Na+占据Li+的位置,表面的Na+不仅起到钉扎效应维持结构稳定性,还可以提高Li+扩散速率,增大倍率性能。表面掺杂的优势在于掺杂离子仅仅进入表面几个原子层的深度,维持表面的稳定性,并且微量的元素掺杂不会对体相材料容量造成较大的损失。但是表面掺杂却忽视了材料体相的结构稳定性。

图9 表面改性
3.1.3 梯度掺杂
结合体相和表面掺杂的优点,人们又发展了一种新的掺杂形式——梯度掺杂,不仅可以维持体相结构的稳定性还能在表面建立保护层等来提高材料的稳定性[54]。Peng等[54]利用共沉淀法实现了Al3+梯度掺杂,得到梯度分布的高镍材料LiNi0.815Co0.15Al0.035O2,元素分布为:Ni 和Co 由内到外呈递减的分布,Al呈现出由内到外为递增的趋势。表面富Al 层对材料表面起保护作用,提高材料的稳定性,1C下1000圈循环后的保持率高达80%。此外,某些元素梯度掺杂会改变表面结构,Pan 等[55]对LiNi0.8Co0.2O2进行Ti4+梯度掺杂,通过原子结构表征及理论分析发现,掺杂后的样品表面形成一层无序层状结构,该表面层抑制了副反应、提高了材料的稳定性。梯度掺杂综合了体相掺杂和表面掺杂的优点,具有表面富元素掺杂和体相少量元素掺杂双重作用,在保证高容量的基础上,提高了表面和体相的结构稳定性。
3.2 表/界面处理
高电压条件下,材料表面与电解液之间有严重的副反应,易被电解液分解产物(HF 等)侵蚀导致结构被破坏,引起材料电化学性能恶化。研究人员通过包覆和表/界面构筑保护层来提高材料表面的稳定性。
3.2.1 包覆
电解液侵蚀材料表面不仅破坏结构还造成过渡金属溶解引发容量衰减,表面包覆可以有效缓解这一问题,提高材料长循环稳定性[56]。包覆剂常用到氧化物[57-60]、氟化物[61-62]、磷酸盐[63-64]等,虽然对材料的性能有一定的提升,但是简单的包覆手段[固相、液相和脉冲激光沉积法(PLD)等方法]难以控制包覆层的均匀性和厚度,有研究表明,非均匀包覆的材料循环后期电化学性能会加剧衰退[65]。为了充分发挥包覆效果,且考虑到富锂锰基材料本身倍率性能差等缺点,Yu 等[66]利用原子层沉积(ALD)法在Li1.13Mn0.517Ni0.256Co0.097O2材料晶粒表面形成一层均匀的、厚度可精准调控的离子导体LiTaO3,如图9(c),并且经过沉积后包覆层和材料表面之间形成一层具有取向生长的均匀的尖晶石中间层,“表面均匀的离子导体包覆层+尖晶石中间层”提供了Li+快速扩散通路,从而提高了倍率性能,且多级表面保护层减小了副反应,增强了循环稳定性,200圈循环后电压降仅为0.9mV/圈。表面包覆技术是最简单且应用最广泛的改性手段,能够有效地稳定材料表面,缓解正极材料/电解液界面副反应的发生。
3.2.2 表/界面改性
对材料表/界面进行修饰能够改变表面的结构从而提高电化学性能[42,67-68]。富锂锰基材料由于Mn4+外层d轨道上的3个电子在八面体场中都占据t2g轨道,远离费米能级故电子难以脱出,所以Li2MnO3被认为是电化学惰性的。阿贡实验室Johnson 等[69]将0.3Li2MnO3·0.7LiMn0.5Ni0.5O2浸泡于HNO3溶液,而后在氨气气氛中热处理,结果表明材料表面脱出部分Li2O形成尖晶石相,改善了首次效率和循环。而后,人们开始对正极材料表面进行处理,使其脱出部分Li2O从而形成保护层。如图9(d),Yu等[42]利用(NH4)2S2O8处理类单晶Li1.2Mn0.567Ni0.167Co0.067O2材料,处理后的材料表面形成尖晶石结相,提高了材料的结构稳定性和热稳定性。Liu等[70]将Li[Li0.144Ni0.136Co0.136Mn0.544]O2在CO2气氛中处理,如图9(e),在该材料表面形成了大量氧空位,抑制了O2释放,提高了稳定性,100次循环后,可逆容量为300mA·h/g 且没有明显的电压衰减。通过表界面改性,在材料表面原位构筑保护层,不仅抑制了与电解液的副反应、稳定了表面结构,同时保护层与基体材料较强的黏附力有效地避免了循环过程中的剥落,从而提高了材料的长循环稳定性。
3.3 掺杂与表/界面改性协同策略
表/界面处理和包覆的主要作用是缓解材料表面与电解液的副反应,但是对材料体相结构的稳定性未达到理想效果。近些年,研究者尝试综合利用掺杂和包覆的协同效应进一步提高材料的稳定性,不仅维持了材料晶体结构的稳定性,还有效地缓解了材料表面与电解液的副反应。Lu等[71]通过理论计算构建模型发现La元素倾向富集在表面、Ti渗入到体相中。以此理论为指导,利用La 和Ti 对高镍正极材料LiNi0.8Co0.1Mn0.1O2进行改性,最终得到Ti掺杂和La4NiLiO8包覆的高镍正极材料LiNi0.8Co0.1Mn0.1O2。Ti元素的引入有效抑制了阳离子混排及循环过程中的相变,包覆层La4NiLiO8不仅有效缓解了与电解液的副反应,而且提高了材料的电子导电性。掺杂和包覆的协同效应使得材料表现出优异的电化学性能。Sun 等[72]利用Mg3(PO4)2包覆Te6+掺杂的富锂材料[见图9(f)]。Te6+的掺杂提高阴阳离子的反应活性和Li+扩散速率,Mg3(PO4)2包覆层抑制了与电解液之间的副反应,且在表面形成的无序结构可以抑制表面氧的活性,维持表面结构的稳定性。此协同效应不仅提高了材料的循环性能(1C下200圈容量保持率为89.2%)和倍率性能(10C时容量为178.8mA·h/g),还降低了电压降。总之,单一的改性手段往往具有一定的局限性,很难达到理想的电化学性能,需要综合考虑多元协同效应的改性手段。
4 总结与展望
本文总结略图见图10。锂离子电池发展至今,提高正极材料的容量仍是最迫切的工作,但随着容量的提高,正极材料结构稳定性和热稳定性差等安全性问题变得更加突出,需要在实际应用中进行改善和平衡,正极材料的未来发展趋势主要有以下几点。
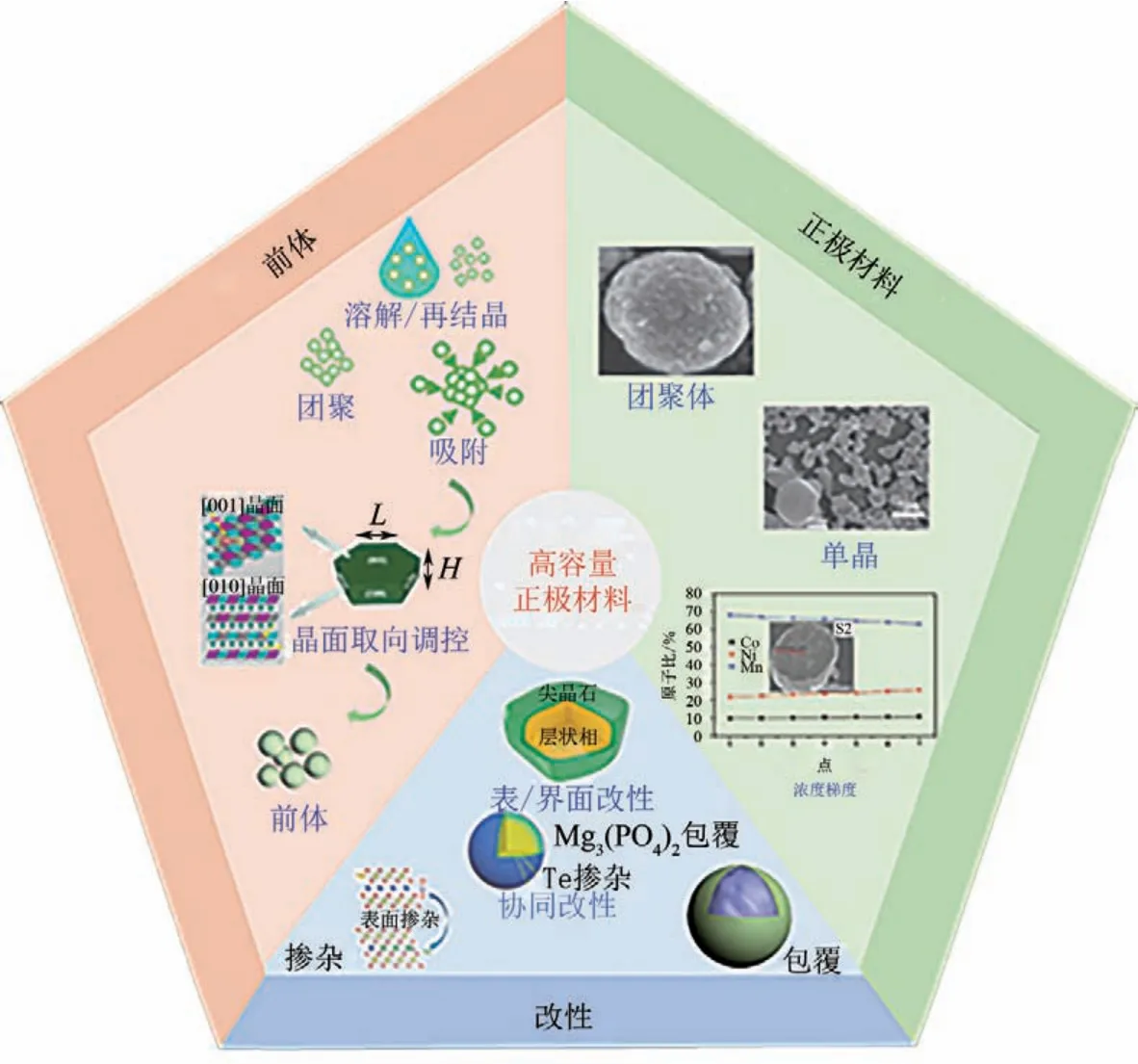
图10 前体合成、正极材料制备及改性
(1)前体工艺的重要性。不管是均一球形还是单晶或梯度材料正极材料的性能主要是由前体的性能决定,因此需要开发出优良的前体合成工艺。在环境和经济效益的双重考虑下,综合考虑pH、氨浓度、反应温度、反应时间等主要影响因素,开发低成本和高可行性的前体合成工艺。
(2)发展性能优异的高容量正极材料。高镍层状氧化物和富锂锰基层状氧化物正极材料具有非常大的潜力,但是这两种材料表现出较差的结构稳定性,明显的电压降(尤其富锂锰基层状氧化物正极材料)等问题。可以通过掺杂、表/界面处理等方法改性,但是往往单一改性手段具有一定的局限性,需要利用多元改性手段的协同效应对材料进行改性。
(3)单晶化及浓度梯度结构设计。虽然能够提高材料性能,但是对于性能提升的机理解释并不清晰,需要借助更加先进的分析手段对其进行研究。
(4)开发兼容的电解液。传统LiPF6为溶质的电解液在高压条件下会发生明显的氧化分解(电压>4.3V时易发生分解)。富锂锰基正极材料的工作电压>4.5V,在此条件下,电解液的稳定性对材料的稳定性具有很大的影响。所以需要开发相匹配的高压电解液,提高电解液与材料的兼容性。
正极材料的容量不断提高,其面临的安全性问题也更加突出,除了对目前主流的正极材料改性修饰外,还应积极探索新型材料来满足不断增长的需求。
猜你喜欢
电子工业专用设备(2022年5期)2022-12-30
中国特种设备安全(2022年4期)2022-07-08
空气动力学学报(2022年1期)2022-03-16
天津医科大学学报(2021年1期)2021-12-05
粉末冶金技术(2021年3期)2021-07-28
空气动力学学报(2021年2期)2021-05-04
科学大观园(2020年22期)2020-11-30
陶瓷学报(2020年5期)2020-11-09
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
