
不同养殖模式下加州鲈池塘的底泥微生物群落结构研究
2021-09-17 12:03姚大龙
中国水产 2021年8期
文/姚大龙
作者单位:河南省驻马店市薄山水库管理局
本试验通过加州鲈混养不同鱼类,投喂不同的饵料,研究底泥微生物的群落结构,为构建健康高效的加州鲈养殖模式提供参考依据。实验于对9个池塘共进行了3次样品采集,经过总DNA提取、16S rDNA序列扩增、高通量测序等,共计获得274786条有效分析序列,经聚类合并共得到4772个操作分类单元(OTU)分类。经分析,在不同养殖和饲料投喂模式下,加州鲈+鲢鳙+黄颡鱼模式下的全饲料投喂组池塘底泥微生物群落更为稳定。
一、前言
微生物是养殖生态系统中的重要组成部分,是有机物的分解者,其在池塘生态养殖中具有重要的作用。外塘养殖中,底泥是分解残饵和碎屑、保证池塘环境健康的重要场所,其微生物组成和功能对于养殖环境的维持非常重要。在不同养殖模式下,养殖环境中的微生物群落会有不同程度的组成差异和动态变化,从而影响养殖生物的生长。因此,探讨不同养殖模式下的底泥微生物的群落组成情况及其差异,有助于了解不同养殖模式的生态学过程,帮助分析不同模式养殖效果产生的原因。
近年来,加州鲈受到广大养殖户的青睐,其养殖产量高、效益好、市场较为稳定,但是随着养殖面积的扩大,养殖中存在的问题也越来越明显。为解决养殖中出现的病害、水质等问题,该品种在饲料投喂方式和养殖模式方面不断升级,由最初的全冰鲜投喂、冰鲜+动物内脏投喂等严重影响水环境健康的投喂方式,逐渐向全饲料投喂转变;由最初的单一养殖,逐渐向多品种混养转变。本实验通过构建9个不同混养模式和投饲方式的组合,分析不同模式下的养殖底泥微生物组成情况,评价不同养殖模式对加州鲈养殖的影响,以期为加州鲈健康养殖模式的推广提供理论依据。
二、材料和方法
(一)实验设计
实验地点位于河南省驻马店市确山县一实验基地进行,共选用标准化池塘9口,该实验田每口池塘的水域面积约为30m×20m,水深1.5m~2m,放养密度约为3000尾/亩。实验历时2个月,于2018年5月22日前完成混养构建,期间采用不同饵料组进行投喂,至7月22日实验结束,将各模式组依次命名为M01-M09,模式构建信息见表1。每日投喂一次,6月22日后至实验结束,每日投喂两次,分别于5月22日、6月22日、7月22日,进行三次样品的采集,采集样品总数为27个。
(二)实验方法
1.样品预处理和总DNA提取
首先称取200mg的样品,放入灭菌的2mL离心管中,加入1mL70%乙醇,震荡混匀,10000rpm室温离心3min,弃置上层液体。将样品管放入55℃烘箱10min,使残留酒精完全挥发,保证后续实验操作。加入1xPBS溶液,震荡混匀,10000rpm室温离心3min,弃置上层液体。倒置2mL管于吸水纸上1min,直至没有液体流出。采用OMEGA试剂盒E.Z.N.ATM Mag-Bind Soil DNA Kit的试剂盒进行总DNA提取。
2.序列扩增
利用Qubit2.0 DNA检测试剂盒对基因组DNA精确定量,以确定PCR反应适宜加入的DNA量。配置好的PCR体系按照如下反应条件进行PCR扩增:94℃变性3min;94℃30s,45℃退火20s,65℃延伸30s,执行5个循环;94℃ 20s,55℃退火20s,72℃延伸30s,执行20个循环;72℃终延伸5min。PCR体系体系按照如下进行:2×Taq master Mix 15μL,Bar-PCR primer F(10μM) 1μL,Primer R (10μM) 1μL,模板DNA 20ng,H20补足至30μL。第一轮PCR所用的引物为细菌16S rDNA V3-V4通用引物:(341F引物:CCCTACACGACGCTCTTCCGATCTG (barcode) CCTACGGGNGGCWGCAG;805R引物:GACTGGAGTTCCTTGGCA CCCGAGAATTCCAGACTACHVGGGT ATCTAATCC)。随后进行第二轮PCR扩增,引入Illumina桥式PCR兼容引物。PCR结束后,PCR产物进行琼脂糖电泳检测,并对扩增得到的DNA产物进行纯化回收。
3.高通量测序及数据质控
采用Miseq测序平台对扩增片段进行双端测序,Miseq测序序列中含有barcode序列,以及测序时加入的引物和接头序列。首先需要去除引物接头序列,再将成对的reads 拼接(merge)成一条序列,然后按照barcode标签序列识别并区分样品得到各样本数据,最后对各样本数据的质量进行质控过滤,去除嵌合体及非特异性扩增序列,得到各样本的有效数据。采用Usearch软件根据序列之间的相似性将序列分成不同的操作分类单元(OTU),通常在97%(属级别)的相似水平下的OTU进行生物信息统计分析。
4.生物学数据分析
对各样本得到的OTU序列进行分类比对鉴定,获得物种信息。对九个实验组的优势类群和优势OUT分类单元进行分析比较,分析各样品的物种组成情况。对样品进行多样性指数分析。采用Shannon index、Simpson index 和Coverage指数评价底泥微生物群落分布多样性。群落结构采用柱状图进行描述,表示各样品中不同细菌操作分类单元所占百分比。采用Chao指数和ACE指数计算微生物群落分布丰度,估计群落中含OTU数目。为了解各样品细菌群落结构的分组相关关系,分别采用聚类分析、PCA分析对群落结构数据进行统计。PCA((principal components analysis)是一种研究数据相似性或差异性的可视化方法,通过一系列的特征值和特征向量进行排序后,选择主要排在前几位的特征值,并通过降维分析获得各变量之间的相互关系的统计方法,最终同样获得可视化的坐标散点图。聚类分析首先对多样性距离矩阵进行层次聚类(Hierarchical cluatering)分析,再使用非加权组平均法UPGMA (Unweighted pair group method with arithmetic mean) 算法构建树状结构,得到树状关系形式用于可视化分析。
三、结果与分析
(一)细菌高通量测序基本情况
经DNA质量检测,九个实验组底泥微生物中均PCR检测到较为丰富的细菌DNA,对九个实验组底泥生物群落多样性开展测序。测序数据经质控、去除嵌合体和非特异性扩增序列,27个样品共计获得274786条有效分析序列,经聚类合并共得到4772个操作分类单元(OTU)分类。各样品的有效序列数和分类单元数等见表2。
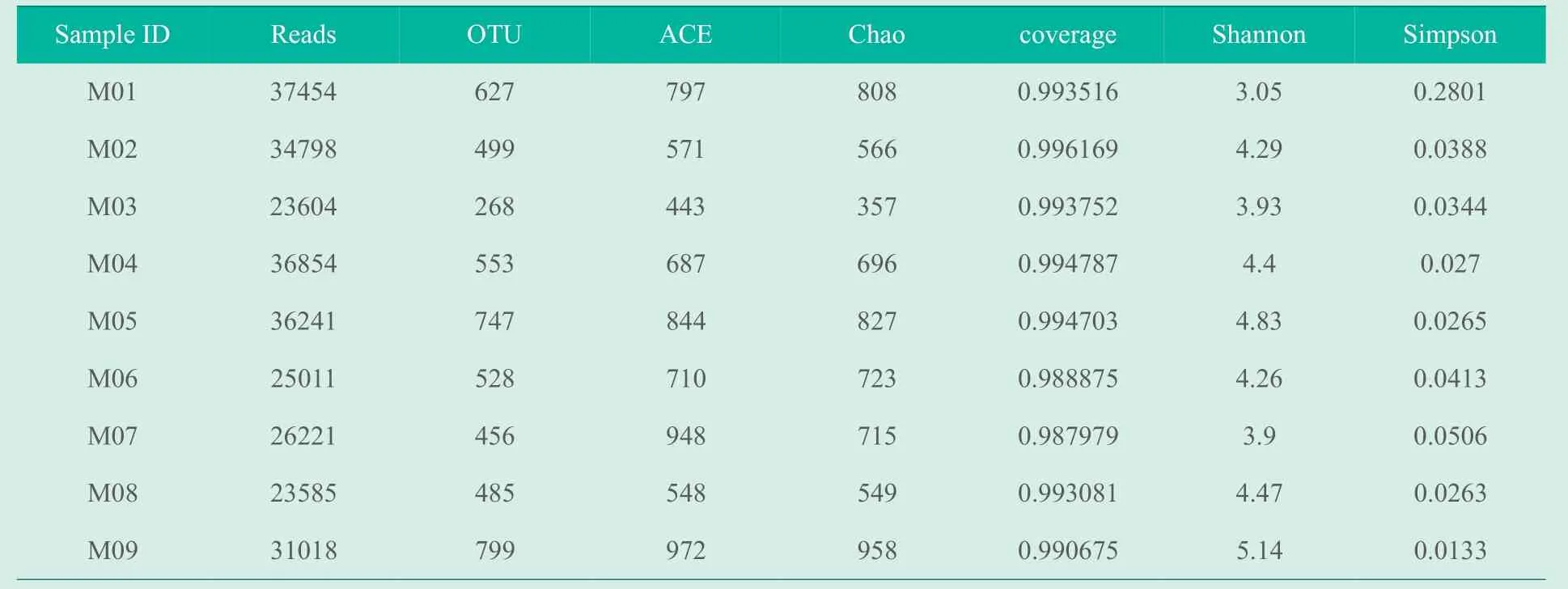
表2 各样品测序分析情况和多样性指数
(二)多样性分析
群落生态学中研究微生物多样性,通过单样品的多样性分析(Alpha多样性)可以反映微生物群落的丰度和多样性。Chao1指数和ACE指数均是用来评价群落中含OTU数目的指数,在本研究中是对底泥微生物群落丰度的反映。
(三)物种组成及优势类群分析
4772个OTU有4098个可以归入变形杆菌门、厚壁菌门、浮霉菌门、拟杆菌门和衣原体门等共五个门当中,其余674个未能确定物种分类归属。分别对九个实验组的OUT按照相对丰度进行排序,发现M01组的底泥微生物细菌群落比例大于1%的细菌优势菌群有11个;M02组大于1%的细菌优势菌群有8个;M03组大于1%的细菌优势菌群有16个;M04组大于1%的细菌优势菌群有13个;M05组大于1%的细菌优势菌群有8个;M06组大于1%的细菌优势菌群有16个;M07组大于1%的细菌优势菌群有16个;M08组大于1%的细菌优势菌群有13个;M09组大于1%的细菌优势菌群有9个。各样品细菌物种组成柱状图见图1。

图1 各样品细菌物种组成柱状图
四、讨论
从本次实验结果可以分析出,各个样品中底泥微生物群落的丰度有很大的差异,其中M09实验组的群落丰度最为稳定,该组为加州鲈+鲢鳙+黄颡鱼全饲料投喂组。
全饲料投喂有利于维持池塘微生物的稳定性,杂鱼的投入有可能造成池塘菌群的剧烈波动,混养模式对底泥微生物的影响没有饲料投喂明显。Shannon指数和Simpson指数显示,M09实验组的细菌群落多样性最高。本研究发现在不同模式下养殖系统的底泥微生物群落结构中变形杆菌门、厚壁菌门、浮霉菌门、拟杆菌门占所有种群的比例最大。采用不同混养模式和投喂模式对加州鲈养殖系统的底泥微生物群落结构的变化均有一定程度的影响,但在加州鲈+鲢鳙+黄颡鱼全饲料投喂模式下,底泥菌群更为稳定,多样性更高,有利于加州鲈的生长和维持池塘底质的生态平衡,符合加州鲈健康养殖模式的要求。
猜你喜欢
农村百事通(2022年10期)2022-11-25
当代水产(2022年8期)2022-09-20
绿色科技(2022年14期)2022-08-12
昆明医科大学学报(2022年2期)2022-03-29
安全与环境工程(2022年1期)2022-02-14
珠江水运(2021年23期)2021-11-23
乡村科技(2021年12期)2021-09-06
汽车观察(2021年11期)2021-04-24
学校教育研究(2020年7期)2020-04-09
焦点(2014年6期)2014-08-19
