
伴放线聚集杆菌感染加重小鼠心肌缺血再灌注损伤*
2021-09-06 14:24:10王冬青
中华老年口腔医学杂志 2021年4期
王冬青 许 晴 任 爽
冠心病及由其引发的急性心肌梗死是全世界死亡及致残的主要原因之一[1]。近年来,我国冠心病的患病率、发病率呈逐年上升趋势。冠心病的治疗临床以血运重建为主。然而,缺血心肌组织突然恢复血流供应后会出现心肌的二次损伤,缺血再灌注(ischemia/ reperfusion,I/ R)损伤心肌可占全部梗死心肌面积的50%[2]。研究显示炎症反应可造成心肌损伤和纤维化,导致心功能进行性损害[3]。几种炎症状态被认为会增加冠状动脉炎症负担。而牙周炎正是一种常见的慢性炎性疾病,以牙齿周围组织的附着丧失,牙槽骨吸收为特征,定植在牙周袋内的以革兰氏阴性菌为主的菌斑微生物引发的一系列炎性免疫反应[4]。
越来越多的研究显示,牙周炎是冠心病的独立危险因素。微生物从发炎的牙周位点持续转移到冠状动脉被认为会导致动脉粥样硬化斑块不稳定,并最终增加急性心肌梗死的风险[5,6]。伴放线聚集杆菌(Aa)是一种革兰氏阴性、兼性厌氧杆菌,包含内毒素、白细胞毒素等多种致病毒力因子,在牙周炎中起重要作用。我们之前的研究指出Aa 在急性冠状动脉综合征(ACS)患者的口腔样本中有发现,而在慢性冠心病患者中没有发现。同时,ACS 患者的血清抗Aa 特异性抗体水平高于慢性冠心病患者[7]。赫尔辛基大学的团队[8]发现Aa 阳性患者中,与“无显著冠心病”患者相比,“稳定的冠心病”和“ACS”患者Aa 水平分别增加了6 倍和2.6 倍。同时“稳定的冠心病”和“ACS”Aa 阳性患者的唾液Aa 抗体水平也显著高于“无显著冠心病”患者。但是,Aa 对心肌缺血再灌注后心室重构的病理影响并不很清楚。因此,本研究的目的是探讨Aa 系统性感染对实验性心肌缺血再灌注损伤的影响。
1.材料与方法
1.1 主要仪器及试剂 小动物呼吸机(Minivent 845,Harvard),气管插管(一次性静脉留置针,型号22G),自制小动物开胸器,超高分辨率小动物超声影像系统(Vevo 770,High Resolution Imaging System,VisualSonicsInc,Canada)。氯化三苯四氮唑(TTC,Sigma),伊文思蓝(Evans blue,Sigma),Masson 三色染色试剂盒(Sigma)。胰蛋白胨-大豆-血清-杆菌肽-万古霉素(TSBV)选择性琼脂固体和液体培养基(自制)。
1.2 实验动物 SPF 级雄性C57BL/ 6(野生型)小鼠,6~8 周,购自北京维通利华实验动物技术有限公司。
1.3 实验方法
1.3.1 小鼠皮下腔室制备及分组 根据之前的实验方法制备皮下腔室[9]。用0.5mm 不锈钢丝制备线圈,并通过手术植入全部小鼠的背部皮下。小鼠随机分为2 组:A 组12 只,皮下腔室内磷酸盐缓冲液(PBS)注射;B 组12 只,皮下腔室内Aa 注射。本研究获首都医科大学附属北京口腔医院动物伦理审查批准。
1.3.2 细菌培养及免疫 Aa ATCC29524 来自北京口腔医学研究所。Aa 于TSBV 琼脂固体培养基微需氧培养7 天,选取菌落传至液体培养基中微需氧培养3 天,液体培养至3代。收集菌液,获取细菌浓度在108-9Colony-forming units(CFU)/ ml左右,最终调整注射浓度为108CFU/ ml。螺旋管埋入2 周后管内注射Aa 活菌0.1ml,每周1 次,共4 次。对照组用PBS 溶液同样处理。最后1 次注射后行心脏手术。酶联免疫吸附测定法测量抗Aa 特异性抗体水平,测量处死时及螺旋管埋入前取血。
1.3.3 皮下腔室内微生物学分析 在小鼠背部腔室内注入Aa 后,第2 天至第7 天均抽取0.1ml腔室内液体,涂于TSBV 培养基微需氧培养,5~7 天后观察菌落的形成。
1.3.4 心肌缺血再灌注模型制备 小鼠称重后用3.6%水合氯醛溶液(剂量:0.1ml/ 10g)腹腔注射麻醉,备皮,碘伏消毒。小鼠仰卧,心电图机针形电极插于小鼠四肢皮下行心电监测,自制拉钩撑开口腔,小型啮齿动物呼吸器插管。在胸骨正中剪开皮肤,在小鼠左侧分开肌层,找到2、3 肋,从第三肋的上缘打开胸腔,暴露心脏及左心室表面血管。用8-0 无损伤丝线活接结扎左冠状动脉前降支(left anterior descending coronary artery,LAD)。结扎前用静脉留置针后的软管,裁成1mm的小环,穿入8-0的无损伤带线缝合,至心脏表面。结扎LAD,心电图的ST 段明显抬高,并且肉眼观察心肌结扎线到心尖这段范围有明显的颜色变白,说明心肌已经缺血。缺血30min 后软管被移出LAD 重新开放,再灌注开始后肋骨到皮肤逐层缝合。待小鼠苏醒四肢有力时,方可拔出气管插管。
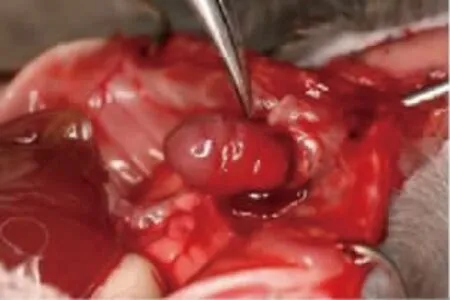
图1 心肌缺血再灌注模型制备
1.3.5 心脏超声心功能评价 实验小鼠仰卧位固定,进行二维超声心动图检查。应用超高分辨率小动物超声影像系统(Vevo 770),RMV-704 单晶片机械扇扫探头,中心频率30MHz。经胸左心室短轴切面获得二维M 型超声图像。测量左心室舒张末期、收缩末期内径与室壁厚度,计算左心室缩短分数(fractional shortening,FS)和心脏射血分数(Ejection Fraction,EF)。以此指标评价各组动物左室功能。
1.3.6 心肌梗死面积评价 TTC-Evans Blue双染:再灌24h 后,将小鼠麻醉接呼吸机维持呼吸,开胸后重新结扎LAD。将带有Evans-blue 溶液0.1ml的注射器,从左心耳穿入,保证针尖注射孔进入左心耳即可。注射伊文思蓝溶液0.1ml。略等2s 后将心脏整体取出,放置-20°冰箱冷冻30min 后切割心脏。一般在结扎部位向心尖处依次约2mm 切割三片。分离右心室,统一切断右心室一侧。随后,放入2%的TTC 溶液染色,保持在37℃15min 观察缺血心肌的存活和坏死区。每片心肌组织称重照相,用Image J 软件计算梗死区(infarct area,IA)、缺 血 危 险 区(area at risk,AAR)、远端区及总面积(左心室面积,LV)。参考文献[10]方法,用IA/ AAR 来评价心肌梗死的面积,用AAR/ LV 评价缺血区面积。每组10 只动物的样本用于统计分析。
1.3.7 病理组织学观察 再灌注24h 后小鼠处死后快速取出心脏,称重后10%福尔马林固定,石蜡包埋,每个心脏获取心尖、中段、心底三个切面行苏木素-伊红(HE)和Masson 三色染色压片,显微镜观察。
1.4 统计学分析 采用SPSS 22.0 专业统计软件处理数据,用Shapiro-Wilk 检验评估数据的正态分布。组间比较用方差分析或t 检验。数据用均数±标准差()表示,P<0.05 为差异有统计学意义。
2.结果
2.1 皮下腔室内液体微生物分析 皮下螺旋管注入活菌后无小鼠死亡,第2~7 天的腔室内液体培养后均可以见到菌落的出现,并成下降趋势。
2.2 血清抗Aa-IgG 抗体滴度 Aa 感染组I/ R小鼠的血清抗Aa-IgG 抗体水平明显高于未感染Aa的对照组I/ R 小鼠(图2)。
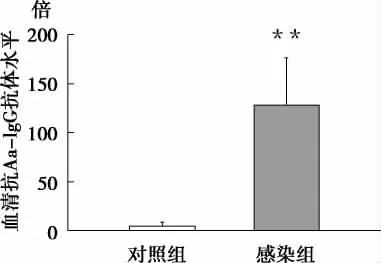
图2 血清抗Aa-IgG 抗体水平
2.3 心脏超声影像检查 图3A 和3B 是对照组I/ R 小鼠(I/ R+PBS)和感染组I/ R 小鼠(I/ R+Aa)左心室M 模式下的代表性超声心动图。如表1所示,Aa 感染的I/ R 小鼠左心室收缩末期内径增大,舒张期末期内径有所减小,但两组间并没有统计学差异。与对照组的I/ R 小鼠相比,感染组的I/ R 小鼠的EF 和FS 明显下降,两组间有显著统计学差异(P<0.01)。
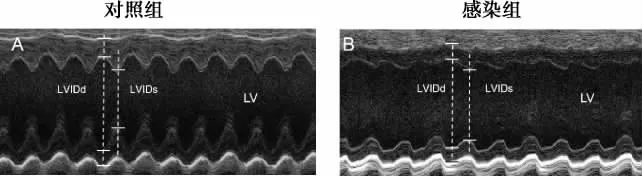
图3 小鼠胸骨旁左心室短轴切面M 型超声图像
表1 感染组和对照组的左室功能比较()

表1 感染组和对照组的左室功能比较()
**,P<0.01,与对照组相比感染组EF(%)和FS(%)明显下降。
2.4 心肌梗死面积 Evans blue-TTC 染色后梗死区呈白色、缺血危险区呈红色和非缺血区呈蓝色(图4A)。图4B 显示与对照组(PBS)组相比,感染组(Aa)小鼠冠脉缺血30min,再灌注24h 后梗死面积/ 危险区面积明显增加(P<0.05),两组间危险区面积/ 总面积具有可比性。

图4 小鼠心肌组织TTC 染色
2.5 组织病理学 Aa 感染组小鼠心肌炎性细胞浸润(图5)和心肌纤维化(图6)相较于对照组未感染小鼠更明显。
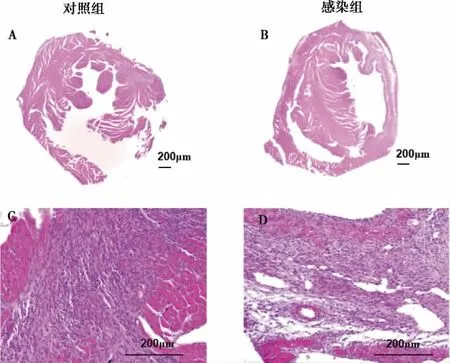
图5 小鼠心肌组织病理图(HE 染色)

图6 小鼠心肌组织病理图(Masson 染色)
3.讨论
中国心血管病患病率处于持续上升阶段,根据生命损失年估计,卒中和缺血性心脏病是我国人群死亡和过早死亡的主要原因[11]。建立稳定有效的小鼠I/ R 模型有助于研究急性心肌梗死和心肌缺血再灌注的损伤和修复。本实验采用常规呼吸机支持改进的心脏原位结扎法[12]制作小鼠心肌缺血再灌注模型,准确性强,操作相对简单,模型较为稳定,术后小鼠的存活率为87.5%(感染组死亡2 只,对照组1 只)。
牙周炎促进全身炎症[13]与冠心病相关联[14,15]。对牙周主要致病菌的高血清抗体水平与冠心病有关[6,14,16]。参照以往经验和方法[9],我们成功建立了皮下螺旋管腔慢性牙周致病菌慢性感染模型。该模型中,管腔内细菌附着于金属管表面,不断向外释放细菌,引起持续性感染。本实验中管腔内可持续监测到Aa 活菌,同时Aa 感染小鼠血清抗Aa 抗体显著升高,表明Aa的感染确实出现在本模型。
以前研究发现,急性冠脉综合征患者血清抗Aa 抗体水平升高[7]。本研究发现Aa 感染小鼠缺血再灌注后,相较于未感染组的小鼠,血清抗Aa-IgG 抗体水平升高的同时,HE 和Masson 染色显示炎性细胞浸润增加,心肌间质纤维化加重。心脏超声显示感染组小鼠左心室室壁变薄,短轴缩短率(FS)及射血分数(EF)下降。这些结果提示Aa 感染加重缺血再灌注后的心肌损伤,牙周炎会影响心肌缺血再灌注后的心室重塑。
Aa 可促进凝血[17],侵入血管内皮细胞[18]。Aa感染引起血清脂多糖和TLR2/ TLR4 表达显著增加[19],活化炎性细胞释放基质金属蛋白酶(Matrix metalloproteinase,MMP)9[20]。MMP 是调节细胞外基质主要成分,不仅能直接降解基质,还能调控心肌梗死后的细胞因子和趋化因子浓度,参与炎症反应,在心肌组织重构中起重要作用[21,22]。在心肌I/ R 损伤动物模型[23,24]和临床患者[25,26]中,均发现MMP-2 和MMP-9的表达增加,且随着损伤时间的延长而增加更加明显。铃木淳一的团队[27]通过皮下螺旋管腔管感染模型发现Aa 通过激活MMP-2引起压力造成的小鼠心肌肥大的恶化。该团队还发现另一重要牙周致病菌牙龈卟啉单胞菌(Porphyromonas gingivalis,Pg)通过CD11b 阳性细胞、细胞因子和MMP-9的表达加重小鼠实验性自身免疫性心肌炎[28]。我们之前的研究[9]发现Pg 通过上调S100A9、MMP-9的表达来促进小鼠实验性腹主动脉瘤的形成和发展。综上所述,伴放线聚集杆菌感染加重小鼠实验性心肌缺血再灌注损伤,MMPs 可能是其中的关键因素,但还需要进一步探讨。
猜你喜欢
昆明医科大学学报(2021年4期)2021-07-23 01:22:00
智慧健康(2021年33期)2021-03-16 05:48:04
中国临床医学影像杂志(2019年1期)2019-04-25 06:49:44
癌症进展(2016年9期)2016-08-22 11:33:18
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:53
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:34
山东医药(2015年14期)2016-01-12 00:39:45
中国医疗美容(2015年1期)2015-07-12 10:06:37
医学研究杂志(2015年4期)2015-06-10 06:42:43
长江大学学报(自科版)(2014年27期)2014-02-27 07:08:27
