
宁东煤热解初始段硫迁移的分子动力学模拟
2021-09-03 08:55梁文政王翠苹
洁净煤技术 2021年4期
王 坤,梁文政,王翠苹
(1.青岛大学 机电工程学院,山东 青岛 266071;2.山东科技大学 土木工程与建筑学院,山东 青岛 266590)
0 引 言
我国作为能源大国,传统的化石燃料,尤其是煤,在热电、工业生产活动中仍将长期占据主要地位。作为CO2、PM2.5等污染物产生的主要源头,燃煤污染防治一直是我国重点关注与控制对象。因此,深入了解煤的结构及其基本反应特征,尤其是从原子尺度揭示其中组成元素在热转化过程复杂的化学演变机理,一直都是煤科学研究领域的核心问题。近些年日益得到认可的分子动力学计算有助于从微观角度解释宏观、复杂的化学反应体系性质,从而为实际应用提供理论基础[1],为煤科学的研究注入了新的活力。
煤的化学结构分析与模型的构建是从分子层面研究煤科学的重点内容,过去几十年间,众多学者通过试验与计算,构建了超过130种不同的分子尺度的煤结构[2]。如今随着科技的进步,分析手段与计算机水平的发展,利用计算机辅助分子设计的方法逐渐兴起。如许多学者利用X射线光电子能谱(X-ray photoelectron spectroscopy,XPS)、13C固体核磁(13C Nuclear magnetic resonance ,13C-NMR)、高分辨率透射电子显微镜(High resolution transmission electron microscope,HRTEM)等分析手段构建的煤化学结构,继而计算获得了与试验数据吻合较好的效果,证明这些手段有效、合理[3-4]。煤化学结构模型的建立使得煤热化学转化研究得以在分子层面展开,为研究其中复杂的化学反应机理提供了机会。如热解是煤燃烧过程复杂且重要的阶段,主要是由自由基驱动的反应阶段,通常分为热解产生挥发物(一次反应)及挥发物的后续反应(二次反应)2个阶段[5]。近些年对该过程的相关试验研究[6-7]较多,但仅通过试验手段难以对煤热解过程中自由基进行检测和分析[8]。因此基于反应力场的分子动力学模拟方法探究煤热解过程应运而生,且发展迅速。
反应力场(Reactive force field,ReaxFF),是一种基于键级的分子力场,常用于分子动力学模拟,由van Duin等和加利福尼亚理工学院合作提出[9-10]。ReaxFF力场中,系统能量以键能、过饱和键能、不饱和键能、键角能、共价键修正能、二面角能、共轭能、范德华相互作用力能以及静电相互作用能之和表达。因此ReaxFF避开了显式的键并基于键级,与传统力场相比,可以满足断裂和形成键的要求,进而可以模拟化学反应而无须预设反应路径。与量子计算(Quantum computation,QC)相比,该方法在考虑化学反应的同时,又可以将模拟体系的规模提高一个量级,与传统分子动力学(Molecular dynamics,MD)模拟相比,该反应力场可连续描述键的生成和断裂且不用预设反应路径。因此ReaxFF反应力场被广泛应用于有机大分子的化学反应动力学模拟领域,目前已参数化并测试了烃的反应、烷氧基硅烷胶凝、过渡金属催化的纳米管的形成和高能材料。在分析煤热解过程方面,Zhan等[11]采用Hatcher次烟煤模型,利用ReaxFF-MD模拟并解释了典型热解产物生成机理,Salmon等[12]模拟了Morwell褐煤模型热解过程中官能团的热解与生成规律。Hong等[13-14]对准东煤的热解过程以及焦油的二次反应原理进行研究。此外,还有采用Wiser等煤分子结构模型,利用ReaxFF-MD模拟褐煤热解机理[15-17]。需要指出的是,目前研究中,虽然对煤热解的经典产物分析较多,但构建的煤化学结构通常只包含C、H、O三种元素,对其中污染性元素如含S的煤化学结构进行构建及S元素迁移的研究相对较少,开展S的转移规律和产物生成路径的深入研究,为含硫污染物的调控和燃烧中控制机理提供新思路。
宁东煤田盛产优质的动力和气化用煤,宁东煤的清洁利用将在振兴宁夏经济,解决西北地区能源供需差距,缓解全国能源紧张状况起重要作用。本文选取宁东煤作为研究对象,通过元素分析、工业分析、XPS、13C-NMR等分析手段,确定其中硫元素的主要存在形式,从而构建了2种主要硫结构形式的低阶宁东煤化学结构。通过Material Studio软件在合适的热解温度下进行宁东煤热解反应的分子动力学模拟,探索热解过程中典型产物以及含硫组分的分布规律,揭示宁东煤热解过程中硫元素的迁移行为。
1 模型构建与模拟方法
1.1 煤样的工业分析和元素分析
为构建宁东煤的化学结构,对宁东煤进行试验表征。试验前将煤粉充分研磨、过筛至75 μm以下待用。采用元素分析仪(型号VARIO EL III)测定煤样碳、氢、氮元素含量,采用全自动定硫仪(型号ZDL-9)测定煤样硫含量,氧元素由差减法计算所得。工业分析参考GB/T 212—2008《煤的工业分析方法》进行。宁东煤工业分析与元素分析见表1,并通过归一化计算处理得到各元素与C元素之间的原子比,结果见表2。

表2 宁东煤样原子比
由表1、2可知,S元素含量很低,由原子比计算可知,在保证所构建的煤化学结构中至少存在一个硫原子时碳原子为300个,构建的宁东煤干燥无灰基化学结构化学式大体为C300H242O64N2S,摩尔质量约为4 931.16 g/mol。

表1 宁东煤样品的燃料分析
1.2 煤样XPS分析
为探究原子间成键方式,对宁东煤样进行了XPS表征测试,仪器为Thermo Fisher公司ESCALAB XI+型X射线光电子能谱仪,测试结果如图1所示。
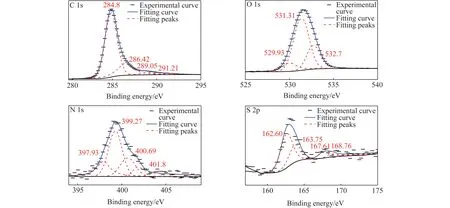
图1 宁东煤样XPS谱图Fig.1 XPS spectra of Ningdong coal
通过Avantage软件对各光电子能谱进行分峰拟合分析,得知宁东煤中C、O、N、S等元素的存在形态及化学环境见表3,峰位信息参考Avantage自带数据库。


表3 宁东煤样C 1s、O 1s、N 1s、S 2p XPS图谱分析数据
1.3 煤样13C-NMR分析
采用13C-NMR技术建立煤平均碳骨架结构,试验仪器为安捷伦Agilent 600M型核磁共振谱仪,所得图谱如图2所示。运用Origin软件进行分峰拟合,其中化学位移0~80为脂肪碳峰,100~165为芳香碳峰,175~200为羰基、羧基峰。所得峰值详细信息及相对含量见表4。

图2 宁东煤样13C-NMR谱图Fig.2 13C-NMR spectra of Ningdong coal
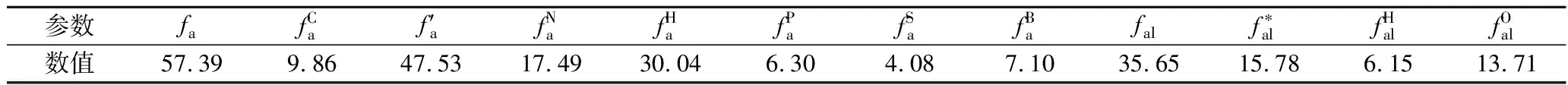
表4 宁东煤样结构参数
根据以上12个峰值数据,通过计算桥碳与周碳比XBP,反映煤中芳环的平均缩聚程度,计算公式为
(1)
其结果为0.175 7,最接近萘的XBP值,为0.25,而蒽的XBP为0.40,得知样品中的芳香结构以苯环和萘环为主,其他芳香结构含量较少。
1.4 模型构建及优化
通过以上分析以及C原子个数,本文选取10个萘环、6个苯环、1个蒽作为基础碳骨架的结构单元,参考文献[4-5,13-14,18]于ChemDraw建立整体宁东煤化学结构如图3所示。其中S原子分别以取代碳链某一羟基中O原子构成R—SH键即硫醇形式,记为模型S1;取代芳香环上某一羟基中O原子构成R—SH键即硫酚形式,记为模型S2。S1、S2将分别进行相同条件的结构优化与热解过程模拟以观察S原子的迁移过程。

图3 宁东煤样化学结构模型Fig.3 Chemical structure model of Ningdong coal sample
一般来说,搭建的初始模型与平衡状态的结构存在较大差别,需进行结构优化以使体系局部能量最小。在Material Studio软件中采用Forcite模块对2个化学解构模型进行循环10次的分子动力学退火模拟,退火温度为500~1 000 K,获得势能面上分子构象优化及能量最小;进行几何结构优化,最终获得稳态几何构型如图4所示。

图4 宁东煤样化学结构3D模型Fig.4 3D chemical structure model of Ningdong coal
利用Amorphous Cell模块,建立2个周期性边界条件的热解模型box(S1)与box(S2),分别包含4个S1结构以及4个S2结构,即每个盒子中含4个硫原子。为消除模型建立过程中原子间可能的重叠区域以及预处理力场与ReaxFF力场势函数的差异,进行ReaxFF MD前,参考文献[10]的方法,首先建立密度较低(0.5 g/cm3)的盒子,后在低温NPT系综下进行密度优化,并最终获得了密度约0.9 g/cm3的初始热解模型如图5所示。
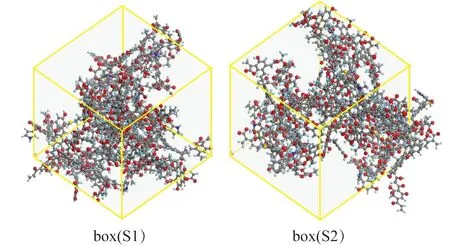
图5 宁东煤样热解初始模型Fig.5 Initial pyrolysis model of Ningdong coal
1.5 ReaxFF动力学模拟
在Material Studio中,利用GULP模块进行分子动力学模拟,力场采用内置的ReaxFF 6.0,系综采用NVT系综,模拟温度为2 500 K,平衡时间为10 ps,时间步长为0.25 fs,模拟总时间200 ps。温度的选取参考冯炜等[4]相关工作,证实在ReaxFF MD中2 500 K 是较为合适的煤热解反应发生的模拟温度。值得注意的是,ReaxFF-MD模拟温度明显高于实际试验温度,其目的是缩短化学反应发生周期,尽管模拟温度与试验温度差异巨大,但模拟文献[15,19]已证实温度评估策略能成功反映试验温度下的反应机理,本文不再赘述。选取总模拟时间时,与本文类似的模拟过程[4,13-14]选取80~250 ps,因此本文选取200 ps作为总热解模拟时间。
2 模拟结果与讨论
2.1 热解产物分布及分析
两热解模型的计算结果如图6所示,其中图6(a)列出了box(S1)的主要热解产物标识,以及硫元素的产物分布情况。由于box(S2)热解情况及主要产物与box(S1)大致相同,这里仅列出硫元素在box(S2)中的主要热解产物形式于图6(b),方便对比。
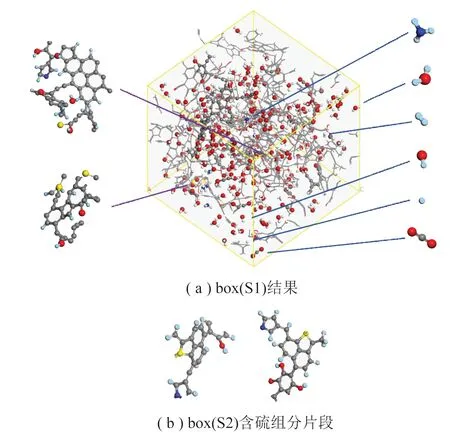
图6 宁东煤样热解模型2 500 K、200 ps时的产物分布Fig.6 Product distribution of Ningdong coal pyrolysismodel at 2 500 K,200 ps
由图6可知,在2 500 K、200 ps的ReaxFF MD热解产物中有大量无机气体分子与活性自由基生成,其中主要为—H、—OH、NH3、水分子、CO2等,此外,还有部分有机气体(含1~4个碳原子)、轻质焦油产物(含5~11个碳原子)、重质焦油产物(含13个碳原子以上)以及焦炭产物(含40个碳原子以上)[20],在该时刻各组分化学式及个数统计见表5。
由表5可知,2个模型的无机气体产物种类相同,各分子数量近似。二者的有机产物在分子数量上都以含1~4个碳原子的有机气体为主,同时有2~3种轻质焦油和多种重质焦油,符合实际试验中煤热解过程释放大量挥发分和产生煤焦油的特点。

表5 宁东煤样在2 500 K、200 ps时的主要热解产物个数
S原子主要集中在重质焦油中,且由于未检测出H2S、COS等含硫气体组分,可推断实际煤热解过程中产生的含硫气体主要产自焦油的二次裂解。
关于键的断裂与产物的生成,通过对轨迹文件分析,发现热解过程中桥键和C—O等键会优先断裂,产生大量活性自由基和中间产物,如—H和—OH,而这些自由基之间的产生与重组会进一步加速热解反应的进行,如可观察到模拟过程中CO2主要来自羧基脱H自由基后进而脱羧形成,这与冯炜等[4]、Zhan等[11]对煤热解过程中CO2形成路径的分析结论一致,而脱下的H自由基和羟基又会成键形成H2O分子。同时热解的焦油产物自身也会进一步释放自由基或与之发生反应,以上这些同样受到二次反应的影响。
为进行粗略定量分析,本文将产物中气体组分以及焦油产物看作挥发分,通过计算2个模型模拟挥发分各组分的分子量总和模拟体系分子量相比得知,box(S1)挥发分析出约25.5%,box(S2)析出约30.3%,表1中Vdaf=27.24%,可以看出2个热解模型的挥发分析出结果均接近试验值,证明所建立的模型具有合理性。同时发现以硫醇形式连接的热解模型更接近实际热解情况,而硫酚形式的连接方式模拟产物焦油组分更多,导致挥发分产出占比偏高。因此综合来看大多数S原子应以硫醇的形式存在。
2.2 硫元素迁移规律
由于不同于C、H、O原子,模型中S原子含量较少(每个模型中4个),无法做到大数量下形态的分布统计,所以本文在整理分析S原子的迁移规律时,只选取某时刻一个S原子存在的形态代表一小段时间内主要的存在形态,所选时刻的硫原子存在形态在系统中至少有2处相同或相似结构,从而总结模拟时间200 ps内硫元素的迁移路径,结果如图7、8所示。
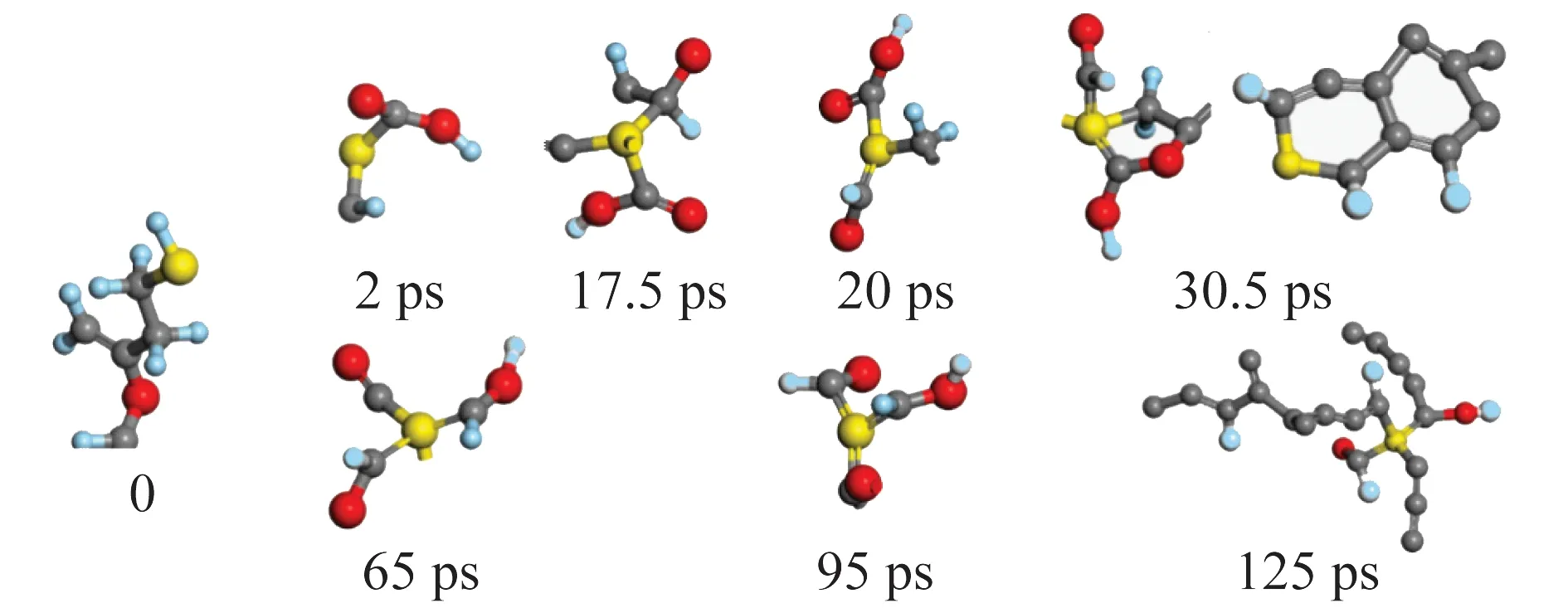
图7 硫元素在box(S1)中的各时间段主要存在形态Fig.7 Main existing forms of sulfur in box(S1)at different time periods
图7中S原子以硫醇形式存在,模拟进行到2 ps时,键能较弱的C—O键首先断裂,在周围各种活性自由基较少的情况下,S—H键断裂,S原子进入到碳链中,形成C—S键;而到17.5~20.0 ps时大多数S—H键开始断裂,且S原子与周围含碳活性自由基或碳链形成C—S键;在30.5 ps时,观察到S、C、O原子之间有成5元环的现象,还趋向于成6元环,而这些环状结构随后会断裂,C—S键变为长链;S原子会继续与C原子成键但不再是环状结构,明显看到重质焦油的生成,多数S原子已迁移到焦油组分的碳链中,为直链形式。到65 ps,4个S原子均与多个C原子成键,直到95 ps均无明显其他结构出现。125 ps至模拟结束,S原子基本只存在于热解的焦油产物中。
因此可认为硫醇热解过程中,S原子更易与多个C原子结合,形成或加入碳链结构,并随之迁移到焦油产物中。
图8中S原子以硫酚的形式存在,模拟进行到10 ps时刻,S—H全部断裂,S原子与周围碳链成键,形成类似噻吩硫形式的环状结构;12 ps时,有部分噻吩硫环状结构在C—S处断裂,S原子与更远处的C原子成键,形成8元环结构。13.5 ps时,S原子开始迁移到焦油物质中,这与以硫醇形式存在的S原子在迁移趋势上相同。

图8 硫元素在box(S2)中的各时间段主要存在形态Fig.8 Main existing forms of sulfur in box(S2)at different time periods
此外这2种环状结构较为稳定,在随后的模拟时间(50.5 ps)中仍长时间存在,直到模拟时间结束,类似噻吩硫的环状结构仍是S原子的主要存在方式。因此推断,当硫酚热解时,容易生成噻吩硫形式的环状结构,且可以稳定存在于热解的焦油产物中。
3 结 论
1)构建的宁东煤C300H242O64N2S化学结构经过退火、几何优化等过程,立体构造显著,层次分明,具有合理性。
2)在ReaxFF MD模拟热解过程中,考察2 500 K、200 ps下的热解效果,发现热解产物中存在大量活性自由基、CO2、H2O、小分子有机气体以及大分子焦油产物,且可以判断活性自由基、产物之间的重组与反应,符合典型热解结果。
3)分析S原子迁移,模型中S原子均易与C原子成键,但S原子以硫醇形式存在时,热解过程中S—H键较易断裂,S原子与多个C原子成C—S键而进入到大分子碳链中,以直链形式稳定存在;而当S原子以硫酚形式存在时,S—H键断裂后,S原子易与碳链成环形噻吩硫结构或8元环结构,也可以稳定存在至模拟结束。
4)S易迁移到焦油产物中,则焦油的进一步热解和燃烧以及S原子的转化途径将在下一步工作中分析,本模拟结果为煤焦油中含硫较高的特性提供理论依据。
猜你喜欢
冶金动力(2022年5期)2022-11-08
选煤技术(2021年6期)2021-04-19
煤矿安全(2020年10期)2020-11-02
同煤科技(2019年5期)2019-11-01
煤(2019年4期)2019-04-28
鞍钢技术(2018年2期)2018-04-13
女士(2017年2期)2017-03-25
化工进展(2015年3期)2015-11-11
中国工程咨询(2012年12期)2012-02-13
