
跨肠道胆固醇排泄的研究进展
2021-09-03 13:10唐晓禹于碧莲综述刘德良彭道泉审校
中国循环杂志 2021年8期
唐晓禹、于碧莲综述,刘德良、彭道泉审校
胆固醇是生物膜上必不可少的组分,也是合成多种生物活性物质的前体物质。胆固醇主要经饮食摄入、自身合成、粪便排泄,在体内维持动态平衡。虽然胆固醇具有重要的生理意义,但是血胆固醇浓度升高却是我国以及世界范围内首位疾病死亡原因——动脉粥样硬化性心血管疾病(ASCVD)的致病性危险因素[1]。在我国,目前高胆固醇血症的患病率高达28.5%,但血脂异常的知晓率、治疗率和控制率整体仍处于较低水平[2]。以胆固醇为切入点控制ASCVD 的方案还有很多进步的空间。胆固醇的分子式是C27H46O,是一种环戊烷多氢菲的衍生物,它可以被转化为生物活性物质,但不能被降解。因此胆固醇排泄对维持胆固醇稳态具有重要作用。胆固醇逆转运(reverse cholesterol transport,RCT)是目前研究较为透彻的胆固醇排泄通路,它清除外周组织中过多的胆固醇,是ASCVD 有力的保护机制。
RCT 对ASCVD 的保护作用源于对动脉粥样硬化斑块内泡沫细胞中胆固醇的清除。泡沫细胞内过载的胆固醇以非特异性液相扩散或受体介导的方式流出细胞,这些受体膜蛋白包括:三磷酸腺苷结合盒转运体A1(ATP binding cassette transporter A1,ABCA1)、三磷酸腺苷结合盒转运体G1(ATP binding cassette transporter G1,ABCG1)和B 类Ⅰ型清道夫受体(scavenger receptor B Ⅰ,SR-B Ⅰ)。流出细胞的胆固醇结合组织间隙中的高密度脂蛋白(HDL)颗粒。HDL 经血液循环到达肝脏,被肝细胞摄取。肝细胞将胆固醇以原型或胆汁酸的形式分泌至胆汁,最后胆固醇经粪便排出体外,这是经典的RCT 模型[3]。在此模型中,肠道仅作为最后一环——胆固醇排泄的通道发挥作用。但近十余年来,越来越多的研究证实,肠道可主动分泌胆固醇至肠腔,直接排出血中的胆固醇,此即跨肠道胆固醇排泄(transintestinal cholesterol excretion,TICE)。在人体,生理情况下此过程占粪便胆固醇排泄总量的35%。
1 跨肠道胆固醇排泄是活跃的生理过程
1959年,Cheng 等[4]发现,胰头癌导致完全性胆道梗阻的患者无胆汁排入肠腔,但其粪便中仍可检出较多胆固醇(约400 mg/d,已校正饮食来源胆固醇),提示存在胆汁以外的胆固醇排泄旁路。1967年,Simmonds 等[5]研究人体肠道胆固醇吸收功能时,对近段空肠灌注,并在相距50 cm 的下游收集灌流液。结果发现,下游位点灌流液中胆固醇含量反而较上游有所增加,提示该肠段本身可分泌胆固醇至肠腔。动物模型证实了该发现,并进一步明确,近段小肠较中、远段小肠分泌胆固醇的能力更强,且该过程依赖于肠腔受体(胆盐、磷脂)的存在[6]。2016年,Jakulj 等[7]则终于在人体实现了对TICE 过程的定量检测。人们从认识TICE 到对其定量分析,经过了数十年。造成研究进展缓慢的原因主要在于胆固醇代谢的复杂性。粪便中胆固醇的来源除TICE 以外,包括三部分:(1)饮食中未被肠道吸收的部分——外源性胆固醇;(2)内环境中的胆固醇,经胆汁排泄进入肠腔,且未被重吸收的部分;(3)随肠腔上皮细胞脱落而丢失的部分(图1)。如何区分不同来源的胆固醇,是研究TICE 时遇到的难点。

图1 粪便中胆固醇的来源
以下多种研究方案巧妙地解决了从整体角度研究TICE 的难题。(1)原位灌注,直接研究单一器官。2007年van der Velde 等[6]向小鼠尾静脉注射同位素标记的胆固醇,30 min 后,原位游离肠段并灌注。在回收的灌流液中可检出标记胆固醇,直接提示小肠可分泌胆固醇至肠腔。(2)外科方式进行胆汁改道,分离胆汁来源胆固醇。De Boer 等[8]以胆道置管的方式,将大鼠1 的胆汁引流至大鼠2 的十二指肠,将大鼠2 的胆汁引流至大鼠1 的十二指肠。胆汁改道后,给大鼠1 腹腔注射含氚标胆固醇的巨噬细胞以检验胆汁途径和非胆汁途径对RCT 的贡献。结果示大鼠1、大鼠2 的粪便中均可检出标记胆固醇,且63%的氚标胆固醇出现在大鼠2 粪便中,37%的氚标胆固醇出现在大鼠1 粪便中,说明TICE 参与RCT 且为旁路途径。(3)遗传方式改变小鼠胆固醇排泄通路上的关键蛋白,在生理状态下阻断胆汁来源的胆固醇进入肠腔。ABCB4 基因敲除、肝脏过表达尼曼-匹克C1 型类似蛋白1(Niemann-Pick C1L1,NPC1L1)[9]的小鼠均表现为胆汁中胆固醇含量显著降低甚至缺如,但以上小鼠粪便中胆固醇含量却无显著下降,同样说明存在肝-胆汁以外的旁路胆固醇排泄途径。(4)人类疾病模型。前述研究方案均不适用于人体研究,但某些疾病本身可造成肝-胆汁排泄通路的完全阻断。类似Cheng 等[4]的研究,Moreau 等[10]给2例同样是胰头癌完全性胆道梗阻的患者静脉注射稳定同位素标记的胆固醇,2 d 后在患者的粪便中发现了标记胆固醇。这个简单清晰的试验提供了TICE 在人体存在的直观证据。(5)同位素标记不同来源胆固醇,直接计算各途径贡献。Jakulj 等[7]给15 名健康受试者口服D4-胆酸,用Entero-test 胶囊行胆汁采样。通过测定胆汁中D4-胆酸含量及胆汁中胆汁酸/胆固醇比值,计算胆汁来源胆固醇。饮食来源胆固醇则以D7-胆固醇标记,并口服不吸收的D4-谷固醇校正粪便排泄量。静脉注射13C2-胆固醇并多次采集血样、粪便,检测13C2-胆固醇的含量。接下来再测定粪便中胆固醇排出量后可计算得到,在生理条件下,TICE 贡献人体粪便胆固醇排出总量的35%。以上研究结果强有力地证明,血浆中胆固醇可跨肠道直接排泄至粪便,TICE 是活跃的生理过程,具有重要的生理意义。
2 跨肠道胆固醇排泄的分子机制
从细胞层面来看,TICE 其实是小肠吸收细胞从基侧面到管腔面的胆固醇流出(图2)。小肠吸收细胞为单层柱状上皮细胞,细胞间的紧密连接将单个吸收细胞的细胞膜分为基底面侧面(基侧面)和管腔面两个功能面。两个功能面上的膜蛋白种类和数量迥异。类似肝细胞,吸收细胞基侧面有低密度脂蛋白受体(low density lipoprotein receptor,LDLR),而管腔面有ABCG5/G8、ABCB1、NPC1L1 等膜蛋白。目前对于TICE 所涉及的分子机制,仍存在许多未解之谜,现对已有证据汇总。
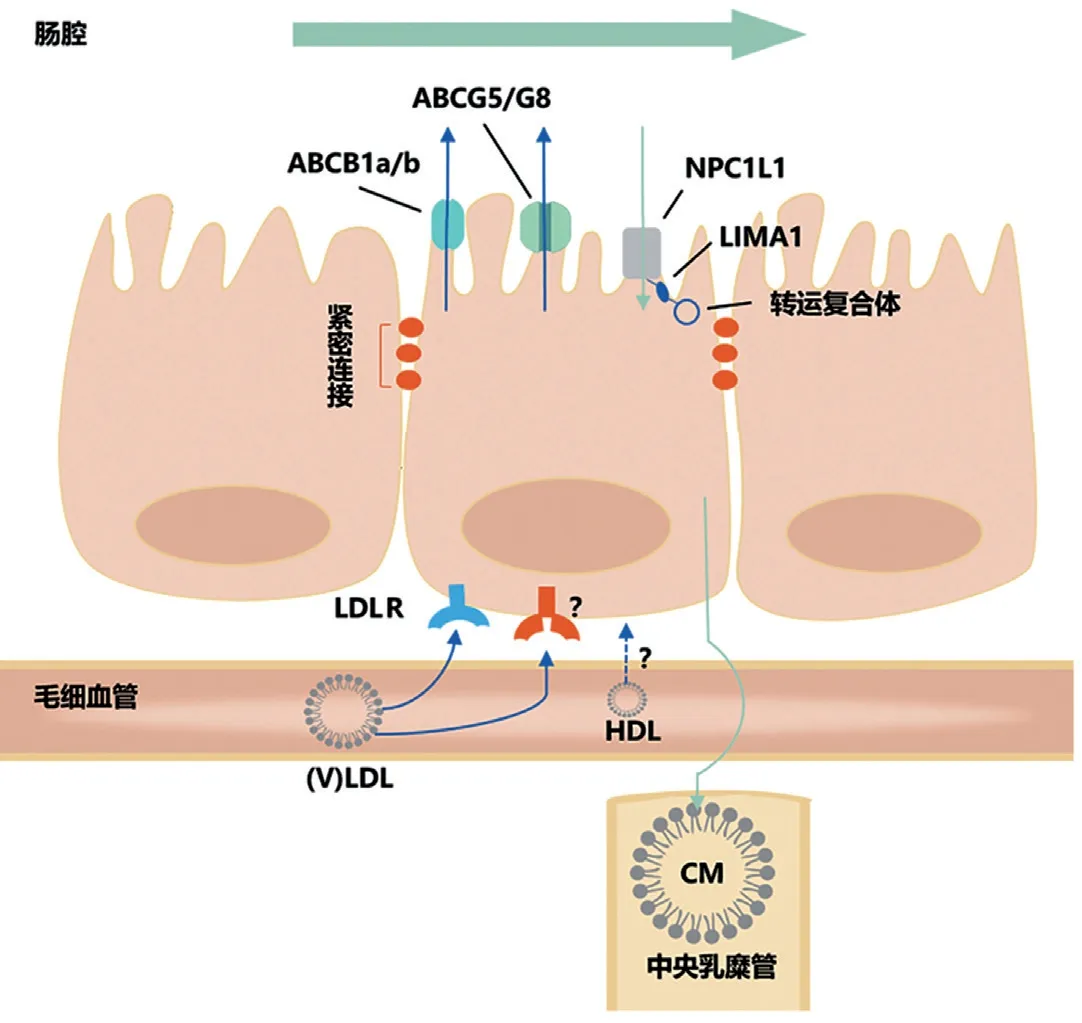
图2 跨肠道胆固醇排泄的分子机制
血浆供体:TICE 是RCT 过程的一环。类比HDL介导肝-胆汁途径的RCT,小肠是否也由HDL 颗粒供给胆固醇呢?目前结论不完全一致。Vrins 等[11]给小鼠尾静脉注射含标记胆固醇的HDL 颗粒并行小肠灌注,结果标记胆固醇基本不出现在小肠灌流液中。但在Le May 等[12]报道的类似实验中,肠腔灌流液可检出标记胆固醇,且基于尤斯室系统的体外实验同样发现HDL 中的标记胆固醇可跨小肠上皮细胞运输。Le May 等认为,造成上述不一致结果的原因可能在于以HDL 为供体的TICE 排出量本身较低,而Vrins 等给药量较少所以检出量亦少。虽然HDL在TICE 中的作用仍有争议,但已经明确含载脂蛋白B(apoB)的脂蛋白是TICE 的供体。在反义核苷酸诱导的肝脏酰基辅酶A:胆固醇酰基转移酶2(Acyl-CoA:cholesterol acyltransferase 2,ACAT2)快速敲除的小鼠模型中,肝细胞内胆固醇酯化障碍,游离胆固醇增多。肝细胞内游离胆固醇通过含apoB 的脂蛋白、经TICE 途径排出体外,引起TICE 增加[13]。若以反义核苷酸敲除肝细胞组装极低密度脂蛋白(VLDL)的关键酶——微粒体甘油三酯转运蛋白(microsomal triglyceride transfer protein,MTP),则TICE 显著下降[14]。以低密度脂蛋白(LDL)为载体的体内、体外实验和对LDLR 的药物干预实验也证实LDL 是TICE 的供体[12]。
基侧面膜蛋白:血浆供体到达吸收细胞基侧面后,是如何入胞的呢?目前已明确的有基侧面膜蛋白LDLR。前蛋白转化酶枯草溶菌素9(PCSK9)可介导LDLR 的降解,减少细胞膜上LDLR 数量。PCSK9 基因敲除的小鼠TICE 显著升高,而PCSK9给药则造成TICE 显著下降,且下降程度在LDLR敲除小鼠中显著减弱[12],说明PCSK9 对TICE 的影响是通过LDLR 发挥作用的,LDLR 是TICE 通路上的一环。但有趣的是在LDLR 基因敲除小鼠,TICE并未消失[12],说明在基侧面除了LDLR,还存在其他受体介导血浆胆固醇入胞,尚需进一步研究以明确该受体。而另一方面,在经典肝-胆汁途径中起重要作用的SR-B Ⅰ在小鼠小肠吸收细胞的基侧面和管腔面均有表达,但对TICE 过程无明显影响[15]。
细胞内胆固醇运输:关于吸收细胞内胆固醇运输的研究较少。在Caco-2/TC7 结肠癌细胞系中,若加入秋水仙碱抑制微管形成,则荧光标记的胆固醇从细胞基侧面向管腔面的转运减少,说明微管参与TICE 途径细胞内胆固醇运输[16]。
管腔面膜蛋白:三磷酸腺苷结合盒转运体G1/G8(ABCG5/G8)异二聚体是管腔面介导胆固醇排出的膜蛋白。肠道特异性敲除ABCG5/G8 的小鼠模型TICE 下降约40%[17]。敲除ABCG5/G8 后,TICE 仍有保留提示ABCG5/G8 并非管腔面唯一介导胆固醇流出的膜蛋白。膜蛋白三磷酸腺苷结合盒转运体B1a/b(ABCB1a/b)可发挥胆固醇翻转酶的作用,介导胆固醇排出。基因敲除或药物性抑制ABCB1a/b 的小鼠TICE 下降26.5%~64.0%[12]。但管腔面是否还存在ABCG5/G8、ABCB1a/b 以外的转运子介导胆固醇排出尚属未知。
跨肠道排出的胆固醇同饮食摄入、胆汁分泌的胆固醇一样,也会被重新收。TICE 排出的胆固醇取决于排出量与重吸收量的差值。事实上,如同肾脏产生大量原尿,肠道也通过TICE 途径排出大量胆固醇,但大量被排出的胆固醇又被重吸收回体内。小鼠模型中,若用药物或遗传方式抑制小鼠对胆固醇的吸收,TICE 可增加至未被抑制时的5~7 倍[18]。在吸收细胞的管腔面,LIM 结构域肌动蛋白结合蛋白1 桥接膜蛋白NPC1L1 和转运复合体上的肌球蛋白Vb,共同协助胆固醇吸收[19]。膜蛋白NPC1L1 是参与重吸收的最重要蛋白,NPC1L1 与ABCG5/G8、ABCB1a/b 共同调控吸收细胞管腔面的胆固醇进出。
3 跨肠道胆固醇排泄的调节
TICE 作为肝-胆汁-粪便途径以外的旁路胆固醇排泄途径,当肝细胞内胆固醇过载或胆汁排泄通路被阻断时,TICE 增加以保证足够的胆固醇排出,维持体内胆固醇稳态。目前已发现,饮食和多种药物可调控TICE。
不同饮食对TICE 的影响不一,肠腔受体的存在显著增加TICE:用不同饮食饲养小鼠后行肠道灌注,高脂肪饮食(无胆固醇,24%脂肪)、西方饮食(0.25%胆固醇,16%脂肪)可分别增加TICE 途径的胆固醇排泄量至普通饮食喂养的2 倍、1.5 倍。而高胆固醇饮食(2%胆固醇,无脂肪)则对TICE 无显著影响[20]。
肝X 受体激动剂显著增加TICE:肝X 受体是配体激活型转录因子,激活后可调控一系列与胆固醇运输相关基因的表达。多项研究已表明,给予小鼠肝X 受体激动剂后,TICE 显著增加[12]。
依折麦布显著增加TICE:依折麦布作用于小肠吸收细胞管腔面的NPC1L1 蛋白,可基本废除肠道胆固醇吸收(胆固醇吸收率从46%下降至4%)[18]。如前文所述,因为抑制重吸收,依折麦布可引起经TICE 途径的胆固醇排泄量大量增加。受试者连续服用4 周依折麦布(10 mg/d),TICE 从(252±46)mg/d增加至(1 024±114)mg/d。
PCSK9 显著减少TICE:PCSK9 基因敲除小鼠TICE 增加62%,而快速注射PCSK9 后,小鼠小肠吸收细胞LDLR 显著减少,TICE 也显著减少约35%[12]。体外实验也发现,给予PCSK9 后,Caco-2/TC7 细胞LDLR 蛋白下降20%,伴随27%的胆固醇流出下降[16]。
不同他汀类药物对TICE 的影响不一:他汀类药物通过竞争性抑制3-羟基-3-甲基戊二酸单酰辅酶A 还原酶,间接上调细胞膜LDLR,发挥显著的降血脂作用。Le May[12]等发现,洛伐他汀使TICE 增加71%。但在另一项研究中[21],只有阿托伐他汀增加TICE,洛伐他汀和瑞舒伐他汀无明显作用。目前尚无在人体中观察他汀类药物对TICE 的影响的研究。
过氧化物酶体增殖物激活受体δ(peroxisome proliferator-activated receptor δ,PPARδ)激动剂减少NPC1L1 的表达而增加TICE:PPARδ 是调节糖脂代谢的转录因子。PPARδ 激动剂可减少肠道NPC1L1 的表达,增加TICE[12]。
法尼醇受体激动剂改变胆汁酸亲水性而增加TICE:法尼醇受体是在肝脏、肠道高表达的转录因子,其配体为胆汁酸。小鼠实验发现,法尼醇受体结合胆汁酸后,刺激吸收细胞释放肠激素成纤维细胞生长因子15 入血,经门静脉到达肝脏,调节肝细胞胆汁酸的合成。法尼醇受体激动剂可通过上述途径显著增加胆汁中亲水性胆汁酸的比例,引起TICE 大量增加[22]。
载脂蛋白A-I(apoA-I)模拟肽4F 显著增加TICE:apoA-Ⅰ模拟肽4F 是人工合成的、功能类似于apoA-Ⅰ的18 氨基酸肽链,具有抗动脉粥样硬化作用。Meriwether 等[23]发现,循环中4F 倾向于到达近段小肠,并跨细胞至肠腔,引起TICE 增加。
4 展望
TICE 作为RCT 中肝-胆汁-粪便以外的旁路胆固醇排泄途径,可在不增加胆结石风险的情况下显著增加胆固醇排泄,降低ASCVD 风险,有重要的药物干预前景。目前,虽然临床已广泛使用他汀类药物、依折麦布调控血脂,但尚无以增加TICE为目标的药物治疗方案。至今TICE 的分子机制还存在很多疑问:是否存在LDL、VLDL 以外的其他血浆供体?吸收细胞的基侧面除了LDLR 以外还有何种受体摄取血浆中的胆固醇?吸收细胞内胆固醇运输的分子机制是什么?管腔面是否还有ABCG5/G8、ABCB1a/b 以外的其他转运蛋白参与TICE 途径?随着这些疑问的逐渐解开,未来可能会出现针对TICE途径的药物治疗方案,在现有药物治疗的基础上更进一步降低血浆胆固醇,降低ASCVD 风险。
综上所述,TICE 是肝-胆汁-粪便途径以外的旁路胆固醇排泄途径。生理状态下,TICE 活跃地将血中的胆固醇泵入肠腔,排出体外,此过程约占人体粪便胆固醇排泄量的35%。当肝-胆汁-粪便途径排泄胆固醇受阻时,TICE 途径的胆固醇排泄量将增加以维持胆固醇稳态。TICE 也可受饮食、药物等多种途径调节。虽然TICE 通路的分子机制还有待进一步阐明,但围绕TICE 的进一步研究有可能为ASCVD 的防治提供新的药物干预靶点。
利益冲突:所有作者均声明不存在利益冲突
猜你喜欢
协和医学杂志(2021年2期)2021-04-16
肝博士(2020年5期)2021-01-18
肝博士(2020年5期)2021-01-18
哈尔滨医药(2016年3期)2016-12-01
海南医学(2016年8期)2016-06-08
创伤外科杂志(2016年12期)2016-02-09
肝博士(2015年2期)2015-02-27
癌变·畸变·突变(2015年4期)2015-02-27
现代检验医学杂志(2015年1期)2015-02-06
中国工作犬业(2015年3期)2015-01-24
