
木犀草素分子印迹固相萃取柱的制备及应用
2021-08-26 10:42:10刘展眉张汉辉东方云肖裕泽程杏安蒋旭红
食品研究与开发 2021年16期
刘展眉,张汉辉,东方云,肖裕泽,程杏安,蒋旭红*
(1.广州南洋理工职业学院教学科研部,广东 广州 510900;2.仲恺农业工程学院化学化工学院,广东 广州 510225)
黄酮类化合物是天然产物中富有活性的植物成分,在医药、食品等领域均有重要的意义。木犀草素是一种具有代表性的天然黄酮类化合物[1],具有抗氧化[2]、抗炎[3]、抗肿瘤[4]以及保护心血管[5]等多种生物活性。花生壳是木犀草素的最重要来源之一[6]。目前木犀草素的分离纯化主要采用有机溶剂提取[7]、硅胶柱层析[8]或大孔吸附树脂吸附[9]等,但由于植物中活性成分种类多样,结构相似,所以存在着分离效果差、溶剂消耗量多以及操作繁琐等缺点,利用传统的分离纯化技术得到高纯度的目标分子存在较大难度[10-11]。因此研究和开发木犀草素的高效分离纯化技术具有重要的意义。
分子印迹固相萃取技术是利用分子印迹聚合物作为固相萃取剂,选择性分离目标物的技术[12]。相较于传统的固相萃取,分子印迹固相萃取对目标化合物具有特异选择性,能分离复杂样品中的特定成分,并且具有较强的富集能力,检测灵敏度高[13]。因此分子印迹-固相萃取现已成为分离纯化的热点之一,有着广泛的应用前景[14-16]。高文姬[17]、肖淑娟等[18]采用本体聚合法制备木犀草素分子印迹聚合物,并以其为填料制备分子印迹固相萃取柱,从花生壳提取物中分离纯化了木犀草素。本体聚合法制备的分子印迹聚合物存在模板分子除去困难、在研磨过程中容易破坏位点、印迹位点效率低等缺点,严重影响了分离柱效及选择性[19],而采用沉淀聚合法制备分子印迹聚合物则不需研磨,颗粒大小较均匀,并且具有更多的均一识别位点[20-21]。
本研究以木犀草素为模板分子,α-甲基丙烯酸(α-methyl acrylic acid,MAA)为功能单体,乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate,EGDMA)为交联剂,采用沉淀聚合法制备木犀草素分子印迹聚合物;并以其为固相萃取柱填料,制备分子印迹固相萃取柱,研究萃取柱对木犀草素的萃取性能,为花生壳中木犀草素的提纯提供了一种高效的分离方法。
1 材料与方法
1.1 材料与试剂
木犀草素(纯度97.81%):上海毕得医药科技有限公司;花生根茎:仲恺农业工程学院种子科学与工程研究所培育;α-甲基丙烯酸(MAA)、乙二醇二甲基丙烯酸酯(EGDMA)、偶氮二异丁腈(azobisisobutyronitrile,AIBN):阿拉丁试剂(上海)有限公司;所用试剂均为分析纯。
1.2 仪器与设备
THZ-82水浴恒温振荡器:金坛市荣华仪器制造有限公司;HH-14数显恒温水浴锅:常州澳华仪器有限公司;KH-45A电热恒温干燥箱:康恒仪器有限公司;ATY124电子天平:日本岛津公司;Agilent-1200S高效液相色谱仪:美国安捷伦科技有限公司;Visiprep DL SPE真空固相萃取装置:美国Supelco公司。
1.3 试验方法
1.3.1 分子印迹聚合物的制备
以木犀草素为模板分子,MAA为功能单体,按1∶8质量比溶解于30 mL甲醇溶液中,加入6 mmol交联剂EGDMA、100.0 mg引发剂AIBN到该混合溶剂中,超声脱气5 min,通氮气5 min脱氧,密封后于60℃恒温水浴中聚合24 h。所得固体以甲醇-乙酸溶液(8∶2,体积比)为溶剂进行索氏提取洗脱,直至洗脱液不含模板分子为止,再用甲醇反复洗涤聚合物除去残留乙酸,真空干燥,得分子印迹聚合物(molecularly imprinted polymers,MIPs)。非分子印迹聚合物的制备除不加入木犀草素模板分子外,步骤与处理方法均与上述制备过程相同,得非分子印迹聚合物(non-imprinted polymers,NIPs)。
1.3.2 高效液相色谱条件(high performance liquid chroma-tography,HPLC)
色谱柱型号:CNW Athena C18-WP(4.6 mm×250 mm,5 μm);流动相:乙腈-水(30∶70,体积比),柱温:28℃,流速:1.0 mL/min,紫外检测波长:350 nm,进样量:20 μL。
1.3.3 木犀草素标准曲线
配制1.0 mg/mL木犀草素标准储备液,甲醇稀释,制备 2、4、8、12、16、20、28、40 μg/mL 的木犀草素标准工作液,按照1.3.2高效液相色谱条件测定峰面积,绘制木犀草素的标准曲线。
1.3.4 木犀草素分子印迹聚合物的吸附性能
1.3.4.1 分子印迹聚合物的动态吸附试验
准确称取30.0 mg MIPs于20 mL具塞锥形瓶中,加入10 mL 1.0 mmol/L的木犀草素标准液,置于25℃恒温水浴中振荡,每隔一段时间(0.5、2、4、5、6、8 h),取出MIPs上清液,通过HPLC测定其中木犀草素的含量,绘制木犀草素吸附量Q(μmol/g)与吸附时间(h)之间的变化曲线。其中吸附量Q按下式(1)进行计算。

式中:Q为吸附量,μmol/g;Co为底物的初始浓度,μmol/mL;Ce为平衡后底物的浓度,μmol/mL;V 为吸附溶液的体积,mL;m为吸附剂印迹聚合物微球的质量,g。
1.3.4.2 分子印迹聚合物的选择性考察
分别称取30.0 mg MIPs与NIPs置于20 mL具塞锥形瓶中,加入10 mL 1.0 mmol/L的木犀草素标准液,置于25℃恒温水浴中振荡吸附8 h,取上清液,利用HPLC检测木犀草素的含量,参照袁波的方法[22],印迹因子IF按下式(2)进行计算。

式中:IF为印迹因子;QMIPs为分子印迹聚合物对木犀草素的吸附量,μmol/g;QNIPs为非分子印迹聚合物对木犀草素的吸附量,μmol/g。
1.3.5 花生壳提取物的制备
称取花生壳粉末8.0 g,加入160 mL乙醇-水溶液(8∶2,体积比),置于80℃恒温水浴中萃取3 h,将得到的乙醇提取液旋蒸至近干得花生壳提取液,自然干燥得花生壳乙醇提取物,密封放入干燥器中备用。
1.3.6 分子印迹固相萃取柱的制备
在固相萃取柱(solid phase extraction,SPE)中通过干法分别装入约0.3 g MIPs与NIPs填料,确保填料紧实平整,再加入少许石英砂于MIPs上方固定。装柱完毕后,加入10 mL甲醇润湿柱子。取2 mL花生壳提取液溶解于不同体积比的甲醇-水溶液后,装入固相萃取柱,以不同体积比的甲醇-水溶液进行淋洗除去杂质,真空抽干,以25 mL不同种类的甲醇-酸溶液进行洗脱,收集淋洗液及洗脱液,HPLC检测其中木犀草素的含量,木犀草素在固相萃取柱中的保留率A、洗脱率B 按下式(3)、(4)进行计算。
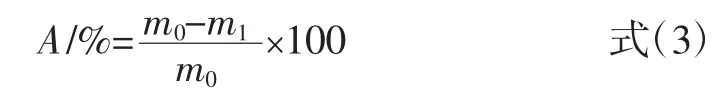
式中:A为木犀草素的保留率,%;m0为上样液中木犀草素的质量,μg;m1为流出液中木犀草素的质量,μg。

式中:B为木犀草素洗脱率,%;m0为上样液中木犀草素的质量,μg;m1为洗脱液中木犀草素的质量,μg。
2 结果与分析
2.1 木犀草素标准曲线
根据木犀草素的浓度C(μg/mL)与色谱图峰面积A(mAU)之间的对应关系,建立线性方程,绘制相对的标准曲线:y=72.026x+19.419,相关系数R2=0.999 2。木犀草素标准曲线见图1。

图1 木犀草素标准曲线Fig.1 The standard curve of luteolin
2.2 分子印迹聚合物制备条件的优化
2.2.1 模板分子与功能单体的摩尔比选择
以木犀草素为模板分子,α-甲基丙烯酸(MAA)为功能单体,固定交联剂EGDMA用量为5 mmol/L,引发剂AIBN用量为100.0 mg,分别以不同摩尔比的木犀草素与 MAA(1∶4、1∶6、1∶8、1∶10、1∶12)制备 MIPs,考察模板分子与功能单体的摩尔比对MIPs吸附量的影响,结果见图2。

图2 模板分子与功能单体的摩尔比对MIPs吸附量的影响Fig.2 Effect of the molar ratio of template to monomer on MIPs adsorption capacity
由图2可知,随着MAA用量的增加,MIPs的吸附量逐渐增加,当木犀草素与MAA的摩尔比为1∶8时,MIPs的吸附量达到最大值32.22 μmol/g,而随着MAA用量的继续增加,MIPs的吸附量逐渐减少。这是因为当功能单体MAA用量较少时,只有少量的木犀草素能与功能单体结合形成复合物,还有大量未结合的印迹分子存在,制备的MIPs中只有较少的木犀草素选择性吸附位点,导致吸附量较少;而随着MAA用量的增加,木犀草素与MAA间形成更多的选择性吸附位点,吸附量逐渐增加,但MAA用量过多时,过量的MAA会导致非选择性的结合位点增加,还会引发自身分子的聚合,导致MIPs中木犀草素的吸附位点减少,吸附量减少。因此,木犀草素与MAA的最佳摩尔比为1∶8。
2.2.2 交联剂用量的选择
固定木犀草素与MAA的摩尔比为1∶8,引发剂AIBN用量为100.0 mg,分别以不同用量的交联剂EGDMA(2、4、6、8、10 mmol)制备 MIPs,考察交联剂用量对MIPs吸附量的影响,结果见图3。

图3 交联剂用量对MIPs吸附量的影响Fig.3 Effect of the cross-linker dosage on MIPs adsorption capacity
由图3可知,随着交联剂EGDMA用量的增加,MIPs的吸附量逐渐增加,当交联剂EGDMA的用量为6 mmol时,MIPs的吸附量达到最大值42.56 μmol/g,而随着EGDMA用量的继续增加,MIPs的吸附量逐渐减少。这是因为当交联剂EGDMA用量较少时,低交联剂用量会使MIPs的印迹空穴刚性过低,洗脱后难以维持空穴原来的形状和大小,导致木犀草素的识别效果不佳,吸附量较少;而随着EGDMA用量的增加,MIPs的印迹空穴刚性增强,减少聚合物在不同溶剂中的溶胀,但当EGDMA的用量过多时,木犀草素在聚合物体系内的传质会被高度交联的网络阻断,导致分离效果下降,吸附量减少。因此,交联剂EGDMA的最佳用量为6 mmol。
2.3 木犀草素分子印迹聚合物的吸附性能
2.3.1 分子印迹聚合物的动态吸附性能
用动态吸附试验考察聚合物MIPs的吸附性能,绘制动态吸附曲线,结果如图4。

图4 MIPs动态吸附曲线Fig.4 Dynamic adsorption curve of MIPs
由图4可知,MIPs在前5 h内吸附量先迅速增大,后缓慢上升,在5 h时MIPs达到最大吸附量37.22 μmol/g,而5 h后,吸附量出现先下降后上升的现象。这是由于在吸附的初期,MIPs的结合位点未达到饱和状态,吸附量随着吸附时间的增加而迅速上升;当吸附量增大到一定程度时,溶液中木犀草素的浓度降低,MIPs出现外吐现象,使得吸附量降低,降低到一定程度时MIPs又重新开始吸附木犀草素。因此,MIPs在5 h时吸附量最大且基本达到吸附平衡。
2.3.2 分子印迹聚合物选择性吸附性能
试验结果表明,在1.0 mmol/L的木犀草素标准液中,MIPs的吸附量可达44.75 μmol/g,而NIPs的吸附量仅有 36.38 μmol/g,印迹因子按式(2)进行计算,得IF=1.23。MIPs比NIPs具有更强的选择吸附性,这是由于NIPs在合成的过程中并没有加入相应的模板分子,无法形成具有特异性吸附的空穴,所以NIPs的吸附主要是物理吸附作用,而合成的MIPs除了具有物理吸附外,还有特异性吸附,因此它的吸附量较NIPs高。
2.4 分子印迹-固相萃取条件的优化
2.4.1 上样溶剂的选择
由于木犀草素在甲醇溶液中溶解性较好,同时花生壳中木犀草素的提取以甲醇-水溶液为溶剂,因此试验分别以不同体积比的甲醇-水溶液(0∶100、30∶70、40∶60、50∶50、60∶40、70∶30、100∶0)作为上样溶剂,考察其对木犀草素保留率的影响,结果见图5。

图5 上样溶剂对木犀草素保留率的影响Fig.5 Effect of sample solvent on luteolin retention rate
由图5可知,随着溶剂中甲醇含量的增加,木犀草素的保留率逐渐下降。当采用纯水作为上样溶剂时,木犀草素的保留率达到最高84.63%,而采用纯甲醇作为上样溶剂时,木犀草素的保留率达到最低56.06%。这是由于木犀草素在甲醇中溶解度较大,当上样溶剂中甲醇浓度较高时,木犀草素容易随着上样液流出萃取柱,造成样品的损失,保留率下降;而甲醇浓度过低时,则溶剂极性过大,容易影响聚合物中印迹空穴与木犀草素物质间的氢键作用[23];且甲醇浓度过低不利于粗提取物的溶解,造成上样过程中部分提取物析出,进行淋洗时,容易随着淋洗液流出,影响萃取效果。故选用甲醇-水溶液(50∶50,体积比)作为上样溶剂。
2.4.2 淋洗液的选择
试验以10 mL不同体积比的甲醇-水溶液(10∶90、20∶80、30∶70、35∶65、40∶60、50∶50)作为淋洗液,考察其对木犀草素保留率的影响,结果见图6。

图6 淋洗液对木犀草素保留率的影响Fig.6 Effect of abluent solvent on luteolin retention rate
由图6可知,随着甲醇含量的增加,木犀草素的保留率呈现下降的趋势。当采用甲醇-水溶液(10∶90,体积比)作为淋洗液时,木犀草素的保留率达到最高98.11%,当采用甲醇-水溶液(50∶50,体积比)时,保留率达到最低39.08%。且当甲醇体积占比大于35%时,木犀草素的保留率快速下降,这是由于甲醇含量的增加,使木犀草素在淋洗液中的溶解度大幅上升,容易随着杂质一起流出,造成保留率骤减。而甲醇-水体积比处于 10∶90~35∶65 时,木犀草素的保留率为 98.11%~90.24%,相对影响较小,因此选用甲醇-水溶液(35∶65,体积比)作为淋洗液。
2.4.3 淋洗液用量的选择
试验以 5、10、15、20、25 mL 的甲醇-水溶液(35∶65,体积比)作为淋洗液,考察其对木犀草素保留率的影响,结果见图7。
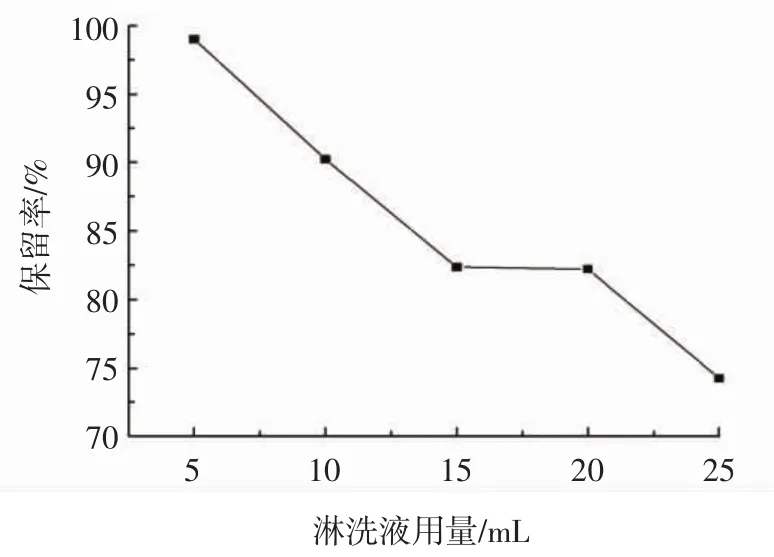
图7 淋洗液用量对木犀草素保留率的影响Fig.7 Effect of dosage of abluent solvent on luteolin retention rate
由图7可知,随着淋洗液用量的增加,木犀草素的保留率呈现下降的趋势。当淋洗液用量为5 mL时,木犀草素保留率达到最高99.00%,而当淋洗液用量为25 mL时,保留率达到最低74.25%。淋洗液用量在5 mL~15 mL时,大部分杂质以及少量木犀草素随淋洗液流出,而当淋洗液用量为15 mL~25 mL时,大量木犀草素开始溶解于淋洗液中,随着淋洗液流出,造成保留率的下降。但是淋洗液用量过低易造成杂质残留。因此选用15 mL的甲醇-水溶液(35∶65,体积比)作为最佳淋洗液。
2.4.4 洗脱液的优化
试验分别选取25 mL甲醇-乙酸溶液(80∶20,体积比)、甲醇-甲酸溶液(80∶20,体积比)、甲醇-乙酸溶液(90∶10,体积比)、纯甲醇溶液作为洗脱液,考察其对木犀草素洗脱率的影响,结果见图8。
由图8可知,当采用纯甲醇作为洗脱液时,木犀草素的洗脱率达到最低67.76%,当采用体积比80∶20的甲醇-甲酸溶液作为洗脱液时,洗脱率达到最高85.57%。这是由于MIPs主要通过氢键作用吸附木犀草素,在保持相同洗脱剂用量的前提下,加入少量的有机酸易于破坏木犀草素与MIPs间的氢键作用,洗脱率上升。甲酸的酸性比乙酸强,所以80%甲醇-甲酸洗脱率最高,因此选用25 mL的体积比80∶20的甲醇-甲酸溶液作为最佳洗脱液。
2.5 花生壳提取物中木犀草素分子印迹-固相萃取纯化
按照1.3.2的HPLC条件对花生壳提取物上样液、固相萃取淋洗液以及洗脱液进行检测,木犀草素标准品、花生壳提取物上样液、固相萃取淋洗液以及洗脱液的HPLC色谱图见图9。液相色谱的分析结果见表1。

图9 HPLC色谱图Fig.9 The HPLC figure
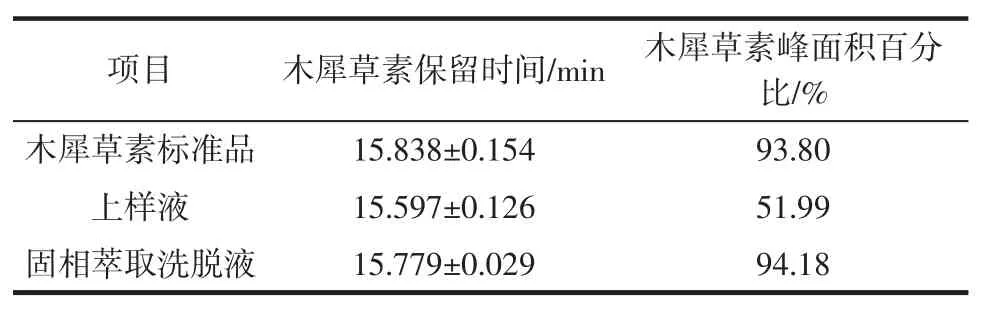
表1 液相色谱的分析结果Table 1 Analysis results of HPLC
由图9、表1可知,木犀草素标准品的平均保留时间是(15.838±0.154)min;花生壳提取物上样液中木犀草素的平均保留时间是(15.597±0.126)min,除此之外还存在大量杂质峰;固相萃取洗脱液中木犀草素的平均保留时间是(15.779±0.029)min,杂峰显著减少。固相萃取淋洗液在保留时间15 min~16 min无木犀草素的特征峰出现,只存在大量杂峰,说明固相萃取的淋洗液可较好地除去杂质。花生壳提取物上样液中木犀草素的峰面积占总面积51.99%,经分子印迹-固相萃取分离纯化后,木犀草素的峰面积占总面积94.18%,提高了42.19%。
3 结论
采用沉淀聚合法制备了木犀草素分子印迹聚合物,该印迹聚合物对木犀草素具有优良的吸附效果以及选择性。以其为填料制备了木犀草素分子印迹固相萃取柱,确定了固相萃取柱的最优上样溶剂为甲醇-水溶液(50∶50,体积比)、淋洗液为甲醇-水溶液(35∶65,体积比)、洗脱液为体积比80∶20的甲醇-甲酸溶液,用此固相萃取柱对花生壳提取物中的木犀草素进行分离纯化,该固相萃取柱对木犀草素表现出较好的选择性吸附效果,可分离纯化出相对纯度为94.18%的木犀草素,为花生壳中木犀草素的分离纯化提供了一种高效的分离方法。
猜你喜欢
Journal of Pharmaceutical Analysis(2020年4期)2020-09-04 09:33:20
中国油脂(2020年3期)2020-04-10 02:08:54
科教新报(2018年48期)2018-06-11 07:19:21
天然产物研究与开发(2018年2期)2018-04-04 02:01:12
中成药(2017年12期)2018-01-19 02:06:56
无机化学学报(2016年8期)2016-12-06 09:05:14
化学分析计量(2016年1期)2016-03-14 00:35:19
广东饲料(2016年8期)2016-02-27 11:10:02
分析测试学报(2015年5期)2016-01-13 06:18:41
分析测试学报(2015年3期)2016-01-13 06:18:20
