
Preparation of Poly(Silicon-phenyl-arylacetylene)Resin by Polymerizing Dialkynylarylenes and Phenylsilane Catalyzed by Alkali Metal Hydroxide
2021-08-25 06:54YANGCanLIUYangFUWeiqiangTONGBinCAIZhengxuDONGYupingSHIJianbing
材料科学与工程学报 2021年4期
YANG Can,LIU Yang,FU Weiqiang,TONG Bin,CAI Zhengxu,DONG Yuping,SHI Jianbing
(1.School of Materials Science and Engineering, Beijing Institute of Technology,Beijing 100081, China;2.Beijing Key Laboratory of Radiation Advanced Materials, Beijing Research Center for Radiation Application,Beijing 100015, China)
【Abstract】 A new method for preparing silicon-containing phenyl arylacetylene resin(SPAR)has been developed using aromatic dialkyne derivatives and 1,4-bis(dimethylsilyl)benzene as raw materials in a one-step reaction in air.In addition, NaOH, a nontransition metal catalyst, was used to achieve high yield polymerization.The experimental conditions affecting polymerization, including monomer species, catalyst dosage, polymerization time, reaction solvent and temperature were investigated.Gel permeation chromatography, nuclear magnetic resonance and Fourier transform infrared spectroscopy were used to determine the molecular weight and structure of the SPARs.Thermogravimetric analysis revealed their thermal stability, and the results indicated that most of them had good thermal stability.This polymerization method that employs simple preparation method, less expensive catalysts, and less complicated posttreatment method will provide an efficient preparation of functional SPARs.
【Key words】 Nontransition metal catalysis;Sodium hydroxide;Silicon alkyne polymerization;Heat-resistant resin
1 Introduction
Polymers containing silicon have many excellent qualities such as good mechanical properties and high temperature resistance[1-2], which allow their broad potential applicabilities in ceramic matrix composites and conductive materials.Silicon-containing phenyl arylacetylene resin(SPAR), a type of silicon-containing polymers, consists of the repeat unit of ―Si―C≡C―Aryl―C≡C―[3], and exhibits after curing many amazing properties, such as high thermal stability and low mass loss under high temperature cauterization[4-5].Therefore, SPARs are widely used as high temperature materials in elec-tronics, information, and aerospace fields[6-7].SPARs have gradually become a hot spot in the field of functional materials in recent years.
The key step in the synthesis of SPARs is the introduction of silicon atoms into the polymers.For example, Harrod utilized amines and copper chloride to catalyze the dehydrogenation coupling reaction between silane and ethynyl compound to obtain SPARs with a yield of 31%-80% in 1990[8].Similarly, Itoh used calcined magnesium oxide as a catalyst to react m-diacetylene benzene with phenyl-silane at moderate temperature between 25-80 ℃ for 2-27 h to obtain solid or viscous liquid polymers with the yield of 40%-96%[9-10].Later, Itoh also synthesized a series of SPARs by condensation reaction between alkynyl Grignard reagent and silane[11-12].In addition, Ueda used Rh(PPh3)3I as the catalyst to prepare a series of poly(silylenevinylene)s in toluene[13].Some of these abovementioned polymerizations need transition metal catalysts, that usually take long synthesis time or generate low yields.
Monomers with triple bonds, such as alkynes and isonitriles, are often used in the development of new polymerizations[14-16].Compared with transition metal catalyzed polymerization, nontransition metal catalyzed polymerization has many advantages, such as easier product separation, higher catalytic effici-ency, mild conditions, low cost, and low biotoxicity[17].Our previous research on the non-transition metal catalyzed triple bond polymeri-zation[18-21]informed us that SPARs could be prepared by an easy-to-get catalyst.TANGetal[22]prepared a conjugated polydiyne at 120 ℃ with three mono-mers, namely, 1,2-bis[4-(iodoethynyl)phenyl]-1,2-diphen-ylethylene, 1,4-bis(2-iodoethynyl)benzene, and 4,4′-bis(2-iodoethynyl)-1,1′-biphenyl, by using potassium iodide as the catalyst and 1,2-dimethox-yethane(DME)as the solvent.Furthermore, GRUBBSetal[23]used NaOH and KOH for the catalytic alkyne and hydrosilane reaction in DME to obtain a series of silicon-containing alkyne compounds.
Based on the preliminary work of our group, we tried to replace the monofunctional alkyne and silane with bifunctional alkyne and silane, and explored the nontransition metal catalyzed polymerization of SPARs according to the aforementioned small molecule reaction[24].In this work, a series of medium/low polymerization degree of SPARs was obtained by one-step polymerization of different diacetylenic monomers with the 1,4-bis(dimeth-ylsilyl)benzene as a hydrosilane, as shown in Scheme 1.This polymerization method can be carried out in air without transition metal catalyst, which makes it cost-effective.Additionally, most of the obtained SPARs showed high thermal stability.Considering its simple synthetic conditions and low cost catalysts, this polymerization technique could provide a new route for preparing functional SPARs.
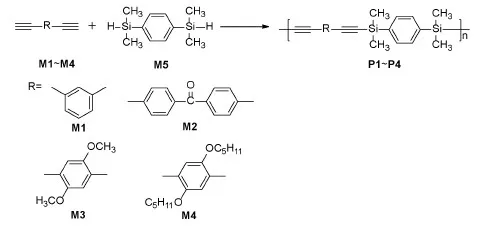
Scheme 1 Synthetic route for SPARs P1-P4
2 Experiment
2.1 Material and equipment
1,3-diacetylene benzene(M1), hydroquinone, 1-bromo-n-pentane, 1,4-bis(dimethylsilyl)benzene(M5), and iodine were purchased from Energy Chemical Co.Ltd.These two monomers were used directly in the polymerization experiment without purification.The other chemicals, like trimethyl-silylacetylene, 4,4′-dibromobenzophenone, 1,4-dibromo-2,5-dimethoxybenzene and tris(4-bromo-phenyl)amine were purchased.
Waters GPC system was used to estimate the weight-average molecular weights(Mw)and polydi-spersity indices(D-=Mw/Mn)of the polymers, which was equipped with a Waters 2414 refractive index detector and Waters 1515 isocratic HPLC pump.Polystyrene were used as standards and THF was the eluent the flow rate of which was 1.0 mL/min.Bruker(ALPHA)spectrometer was used to measure the FT-IR spectra.Bruker AV 400 spectrometer was used to measured1H NMR and13C NMR spectra.CDCl3was the solvent and trimeth-ylsilane was the internal standard.Thermogravi-metric analysis(TGA)was carried out on a DTG-60H.The atmosphere was nitrogen.The heating rate was 10 ℃/min.
2.2 Synthesis of monomers M2-M4
Monomers M2 and M3 were synthesized by using a similar method, but with some adjustments according to the reported references[25-28].We used M2 as an example for introducing the synthetic route(Scheme 2).
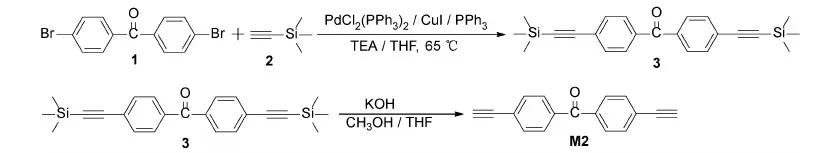
Scheme 2 Synthetic route for M2
4,4′-dibromobenzophenone, the compounds 1(6.8002 g, 20 mmol), PPh3(262.0 mg, 1 mmol), CuI(95.0 mg, 0.5 mmol), PdCl2(PPh3)2(356.0 mg, 0.5 mmol), TEA(150 mL), and THF(25 mL)were added into a 500 mL flask under nitrogen atmosphere.The mixture was heated to 65 ℃ under stirring condition.After about 30 min, the(trimethylsilyl)acetylene 2(8.48 mL, 60 mmol)was added into the flask.After 24 h, product 3 was separated by flash column chromatography.The yield was 84%.
Product 3(1.8701 g, 5.5 mmol), KOH(560.0 mg, 10 mmol), THF(50 mL)and methanol(50 mL)were added into a 250 mL flask, and stirred at room temperature for 12 h.The organic layers were combined and washed with brine and water, extracted, separated, and dried over anhydrous MgSO4for about an hour.The product M2 was separated by flash column chromatography.The yield was 86%.1H NMR(400 MHz, CDCl3): δ=8.62-8.60(m, 4H), 7.63-7.61(m, 4H), 4.07(s, 2H).13C NMR(100 MHz, CDCl3): δ=194.98, 137.06, 132.10, 129.88, 126.52, 82.75, 80.39.HRMS(EI, m/z)Calcd for[M+H]+C17H10O: 230.0732, Found: 230.0731, error:-4.3×10-8.
M3 can be synthesized using a similar procedure as M2(Scheme 3).1H NMR(400 MHz, CDCl3): δ=6.98(s, 2H), 3.86(s, 6H), 3.41(s, 2H).13C NMR(100 MHz, CDCl3): δ=154.40, 116.15, 112.63, 82.81, 79.66, 56.40.HRMS(EI, m/z)Calcd for[M+H]+C12H10O2: 186.0681, Found: 186.0679, error:-1.07×10-7.
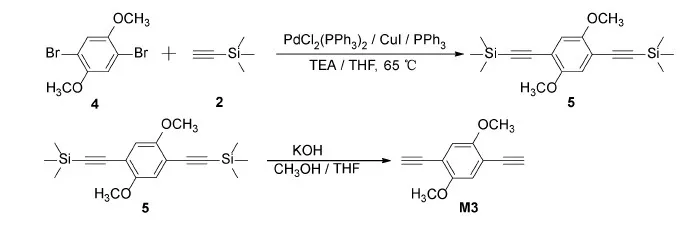
Scheme 3 Synthetic route for M3
The synthesis of monomer M4(Scheme 4)was slightly complicated.Hydroquinone 6(5.5505 g, 0.05 mol), bromo n-pentane 7(22.6575 g, 0.15 mol), acetone(200 mL), and K2CO3particles(13.8205 g)were added into a 500 mL flask.The mixture was heated to 50 ℃ under stirring condition.The reaction lasted for 12 h, then suction filtration and spin-drying were carried out.The product was recrystallized to give product 8 with the yield of 73%.The product 8 was then reacted with iodine to give product 9 with the yield of 87%.Monomer M4 can be obtained by performing a similar procedure such as synthesis of M2.1H NMR(400 MHz,CDCl3): δ=6.95(s, 2H), 3.99-3.95(t,J=6.4 Hz, 4H), 3.33(s, 2H), 1.82-1.79(t,J=6.4 Hz, 4H), 1.47-1.37(m, 8H), 0.94-0.91(t,J=7.2 Hz, 6H).13C NMR(100 MHz, CDCl3): δ=154.00, 117.77, 113.29, 82.45, 79.77, 69.66, 28.84, 28.09, 22.41, 14.03.HRMS(EI, m/z)Calcd for[M+H]+C20H26O2: 298.1933, Found: 298.1925, error:-2.68×10-7.
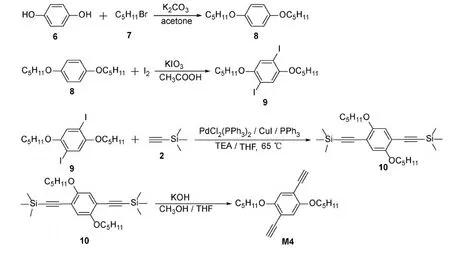
Scheme 4 Synthetic route for M4
2.3 Synthetic procedure of SPARs
In a test tube, 5 mmol of diacetylenic monomer, 5 mmol of M5, 1 mmol of strong base as catalyst, and 5 mL of solvent were added.Then, the reaction mixture was heated while stirring to a fixed temperature for a certain period of time.Let the mixture stand for a while until it cools to room temperature.Then we diluted the mixture with 5 mL of dichloromethane and then added 200 mL of petroleum ether while stirring to obtain the precipitate.Then the precipitate was filtered and dried in vacuum oven at 50 ℃.Finally, SPARs were obtained.
3 Results and Discussion
3.1 Polymerization
The synthesis of all monomers and its charac-terization data are presented in the above experi-mental sections and some original spectra are provided in Figures S1-S6 of the Supporting Information(SI).Monomer M1 was selected as a sample for exploring the optimal polymerization condition, since it could be purchased directly.
First, the effects of catalyst type and amount on the polymerization efficiency were determined under the same experimental conditions(Table 1), i.e., DME was used as the solvent, the reaction proceeds for 12 hours at 80 ℃, and the monomers concen-tration was 1.0 mol/L.Two catalysts of KOH and NaOH were tried in these experiments because both of these are inexpensive and easily available.When the dosage of KOH was very low(only 1 mol% of the monomer), no polymer was obtained(Table 1, No.1).When the dosage of KOH was up to 5 mol% of the monomer, a few polymers were obtained with a very low yield(Table 1, No.2), nevertheless, the polymers showed good solubility in tetrahydrofuran(THF).As the amount of KOH continuously increasing, the yield of the polymers increased but the solubility decreased(Table 1, Nos.3 and 4).Therefore, KOH was found to be unsuitable for the polymerization.When the catalyst was changed to NaOH, a weaker alkaline than KOH, the polymer could be obtained when the dosage of NaOH was 10 mol% of the monomer.However, the yield of polymers was low(Table 1, No.5).When the dosage of NaOH was 30 mol% of the monomer, we obtained the polymer with the highest molecular weightMwand broadD-.However, the yield of the obtained polymer was not high(Table 1, No.7).Meanwhile, when the dosage of NaOH was 20 mol%, the yield of the polymer was good with a mediumMwandD-(Table 1, No.6).Therefore, NaOH was chosen for the next polymerization, and the amount of NaOH was 20 mol% of the monomer.
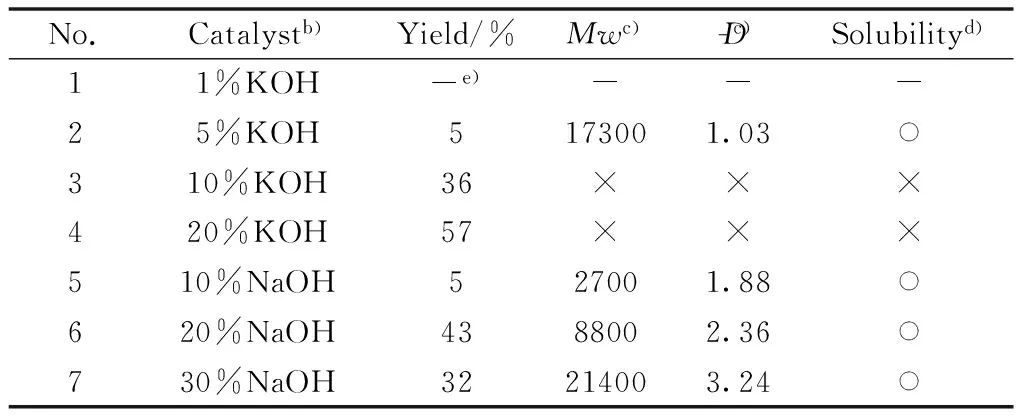
Table 1 Effect of the type and amount of catalysts on the polymerizationa)
Second, the solvents used in the polymerization, such as THF, 1,2-dichloroethane(DCE), and DME, were chosen to investigate the better solvents(Table 2).No polymers were obtained in THF or DCE(Table 2, Nos.1 and 2).The polymerization only proceeded in DME as the solvent(Table 2, No.3), which agreed well with the previous report that DME was a better solvent for small molecules reaction[24].It indicated that highly polar solvent facilitates the polymerization.Therefore, DME was used for the polymerization solvent in the following experiments.
Third, the polymerization time was investi-gated, and Table 3 showed the results.The polymeric product was obtained within only 6 h(Table 3, No.1), but the yield was as low as 22%.When the polymerization time was 12 h, both the yield andMwof the polymer improved(Table 3, No.2).When the reaction time was increased up to 24 h, the yield of polymeric product increased but the obtained polymers were partially soluble(Table 3, No.3).Therefore, 12 h was chosen as the optimal reaction time.
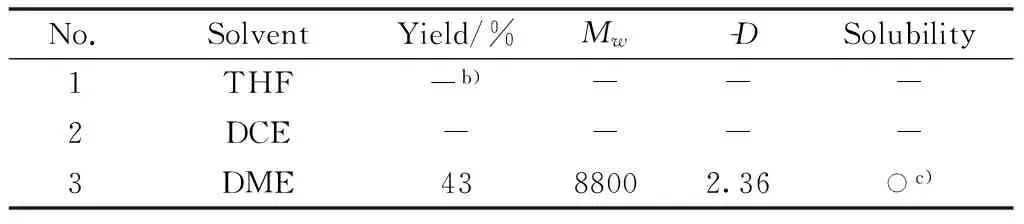
Table 2 Effect of solvents on polymerizationa)
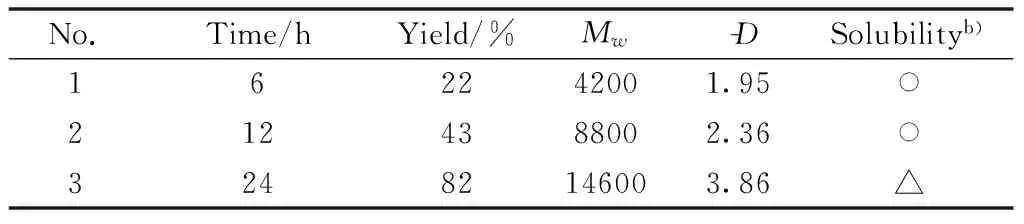
Table 3 Polymerization timea)
Fourth, Table 4 showed the effects of reaction temperature on the polymerization.The reaction temperature was controlled in the range between 60 ℃ and 80 ℃, and the yield of the polymeric product gradually increased with temperature increase.However, the solubility of the obtained products, which were polymerized at 100 ℃, became partially soluble(Table 4, No.3).Therefore, the optimized polymerization temperature was 80 ℃.
Fifth, the effect of the monomer concentration was investigated and the results are summarized in Table 5.Results clearly showed that monomer concentrations had important effects on yield andMw.When the monomer concentration was as low as 0.5 mol/L, the polymer yield was only 17%(Table 5, No.1).The yield of 43% was obtained by increasing monomer concentration from 0.5 mol/L to 1 mol/L and the product had higherMwthan the others(Table 5, No.2).As monomer concentration increased, both the yield andMwdecreased(Table 5, Nos.3 and 4).Under low monomer concentration conditions, the probability of molecular collision is low, which results in low yield andMw.However, the viscosity of the polymerization system would be increased with increasing monomer concentration, which makes it difficult to grow into polymer chains[21].Therefore, both low or high monomer concentrations were unfavorable for the polymeri-zation and the optimized monomer concentration is approximately 1 mol/L for this polymerization system.
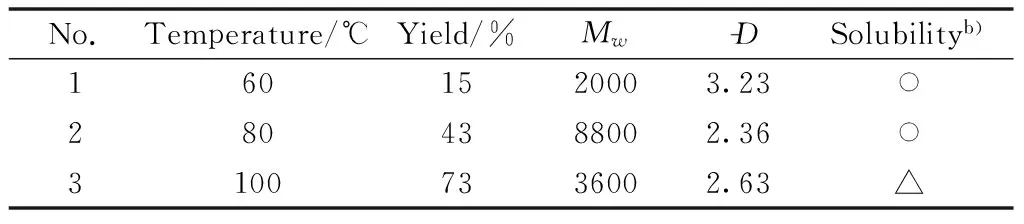
Table 4 Effect of temperature on the polymerizationa)
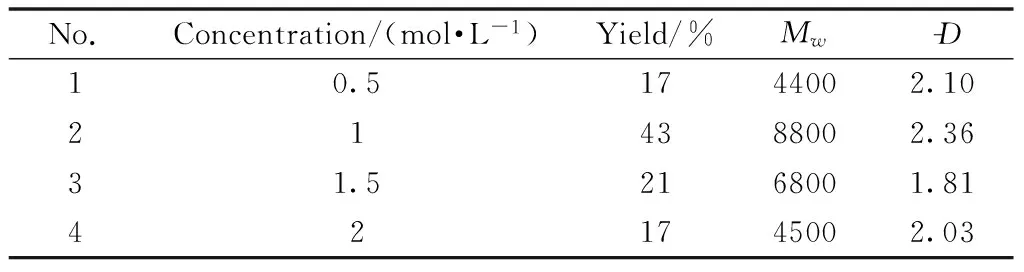
Table 5 Effect of the monomer concentration on the polymerizationa)
Finally, the effect of monomer ratio on the polymerization was investigated under the above-mentioned optimized conditions, and Table 6 showed the results.The ratio of M1 to M5 was changed from 1∶0.8 to 1∶1.2.All the polymeric products had good solubility in THF and moderateMw.The ratio of M1 to M5 at 1∶1 showed higher yield than the other, which is understandable because the equivalent functional groups facilitate the condensation polymerization.Therefore, the ratio of 1∶1 is the optimal polymerization condition.
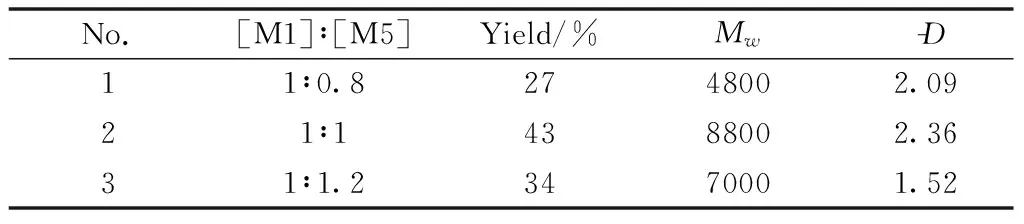
Table 6 Effect of the monomer ratio on the polymerizationa)
The push-pull electron ability of the substituent in the diacetylenic monomers has a great influence on the polymerization[20-21].Under the optimized polymerization conditions previously explored, the scope of diacetylenic monomers was investigated by using M2 that included an electronic-withdrawing group of carbonyl, M3 that included an electronic-donating group of methoxyl, and M4 that included an electronic-donating group of pentyloxyl, and their chemical structures were previously shown in Scheme 1.In Table 7, we summarized the polymerization results.All the obtained polymers showed median yield and good solubility in THF.This polymeri-zation was tolerable to different diacetylenic monomers whether there was an electronic-withdrawing or electronic-donating group.It is worth noting that P4 exhibits a viscous liquid state due to its long alkyl side chains, which is good for the potential applications such as fiber reinforced materials and spin-coating protective coats.
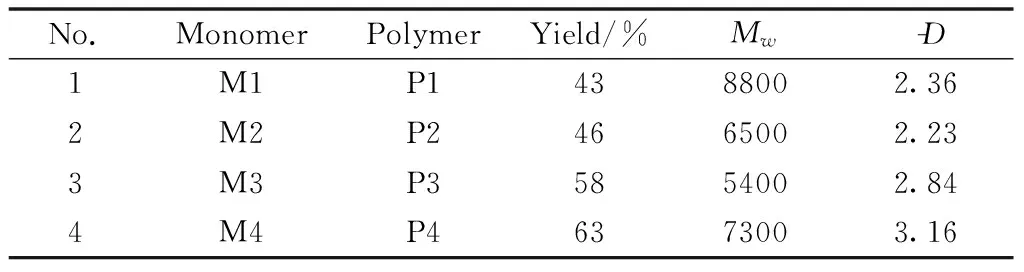
Table 7 Polymerization data for monomers M1-M4a)
3.2 Structural characterization
To confirm the structure of the obtained polymers, the1H NMR spectroscopy of the monomers and polymers were measured.Polymer P1 was taken as an example and shown in Fig.1.The peak atδ=3.09 was assigned to the resonance of Hain M1.The peaks atδ=4.43 andδ=0.33 were ascribed to the resonances of Hband Hcin M5.In polymer P1, all these Hc′still existed with different peaks due to the polymeric polydispersity.There were also tiny peaks of Ha′and Hb′in polymer P1, which indicated that the end group of H-C≡ and H-Si was left after polymerization, mainly due to relatively low molecular weight.All these results indicated that the polymerization proceeded according to its target route and SPARs were obtained by this catalyst system of alkali metal hydroxide.The other similar results were obtained in the1H NMR spectra of P2-P4 and shown in Figures S7-S9 of the SI.
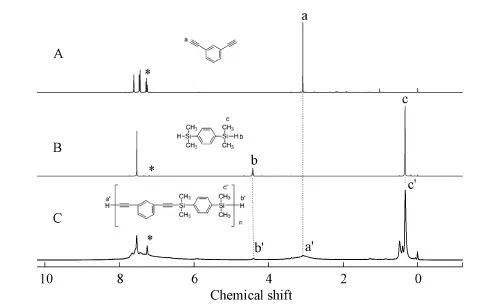
Fig.1 1H NMR spectra of(A)M1,(B)M5 and(C)P1 in CDCl3.The solvent peaks are marked with asterisks
More structural information about SPARs could be obtained from FT-IR spectroscopy.The FT-IR spectra of M1, M5 and P1 were given in Fig.2 as an example.The peak at 3292 cm-1was associated with the stretching vibration bands of H-C≡ in M1, which greatly reduced its intensity in P1.The peak at 2100 cm-1was associated with stretching vibration of C≡C in M1, and it became more obvious after the polymer was formed.The peak at 2143 cm-1was associated with the Si-H stretching vibration in M5, which was weakened in P1.The bending vibration peaks inside the surface at 1251 and 1386 cm-1and the asymmetric stretching vibration peak at 2962 cm-1in M5 that all belonged to Si-CH3could be observed in P1.All these FT-IR results indicated that P1 was the target polymer that had the desired structures.Other FT-IR spectra of P2-P4 also showed similar results(Figures S10-S12 in the SI).
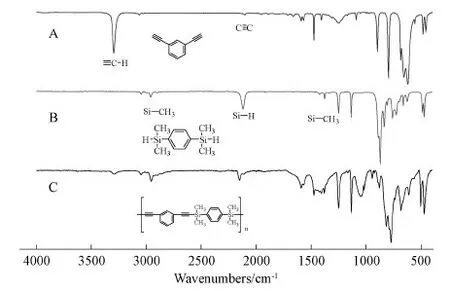
Fig.2 FT-IR spectra of(A)M1,(B)M5 and(C)P1
3.3 Thermal Stability
The thermal stability of P1-P4 was measured by TGA and is shown in Fig.3.The first decomposition temperatures(Td, 5% loss of their weight)ranged from 337.1 to 397.5 ℃ for these SPARs.Among them, P2’s Tdwas as high as 397.5 ℃.Most SPARs showed high carbon residues when the temperature reached 800 ℃.Due to the long alkyl chain in its structure, P4 showed low carbon residues, only approximately 39.4%.The residual carbon content of polymer P2 at 800 ℃ was the highest among these polymers, reaching approximately 75.6%.All these results clearly illustrated that SPARs could be used as heat-resistant coatings or ablation-resistant materials for heat protection.
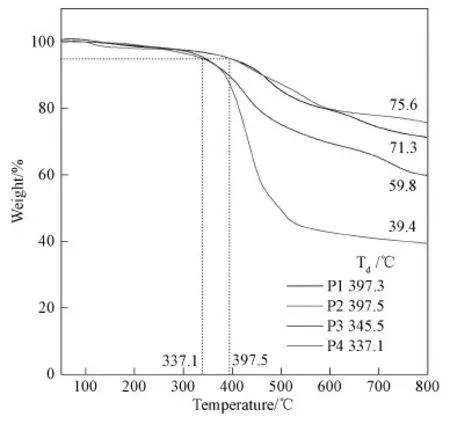
Fig.3 TGA curves of P1-P4 measured under N2 at a heating rate of 10 ℃/min
4 Conclusion
In summary, a synthesis method for SPARs has been developed by utilizing an alkali metal hydroxide catalysis system.Without using transition metal catalysts, the current polymerization method would be cost-effective route for synthesizing SPARs by using NaOH to catalyze a one-step polymerization between aromatic diacetylenic monomers and phenylsilanes.Overall, the following are the best polymerization conditions: 20 mol% NaOH is used as the catalyst, the functional group ratio of monomers is 1∶1, and the reaction is carried out at 80 ℃ for 12 h in 1 mol/L DME solvent.In addition, all obtained SPARs show good thermal stability and have high carbon residual yield up to 75.6% at 800 ℃.Further work focused on the applicability of SPARs is currently under way in our lab.
SupportingInformationfor
1.Structural characterization of monomers
1.1 Structural characterization of M2
13C NMR(100 MHz, CDCl3): δ=194.98, 137.06, 132.10, 129.88, 126.52, 82.75, 80.39.
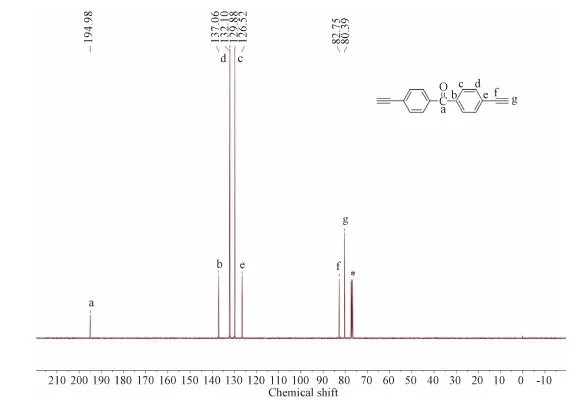
Figure S1 13C NMR spectrum of M2 in CDCl3.The solvent peaks are marked with asterisks
HRMS(EI, m/z)Calcd for[M+H]+ C17H10O: 230.0732, Found: 230.0731, error:-0.43.
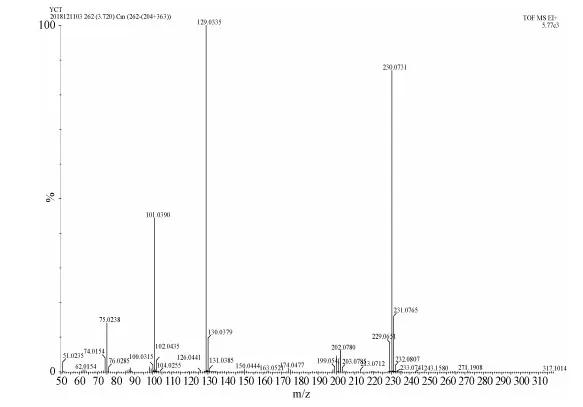
Figure S2 HR-MS spectrum of M2
1.2 Structural characterization of M3
13C NMR(100 MHz, CDCl3):δ=154.40, 116.15, 112.63, 82.81, 79.66, 56.40.
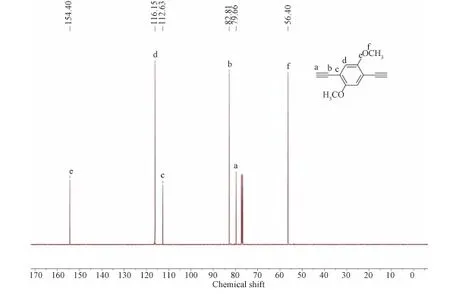
Figure S3 13C NMR spectrum of M3 in CDCl3.The solvent peaks are marked with asterisks
HRMS(EI, m/z)Calcd for[M+H]+ C12H10O2: 186.0681, Found: 186.0679, error:-1.07
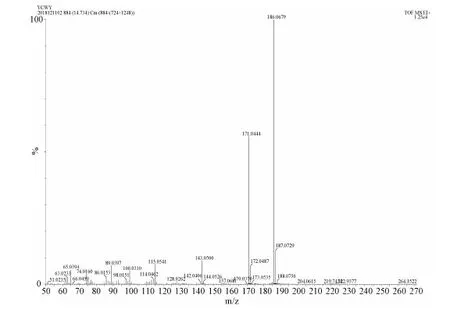
Figure S4 HR-MS spectrum of M3
1.3 Structural characterization of M4
13C NMR(100 MHz, CDCl3):δ=154.00, 117.77, 113.29, 82.45, 79.77, 69.66, 28.84, 28.09, 22.41, 14.03.
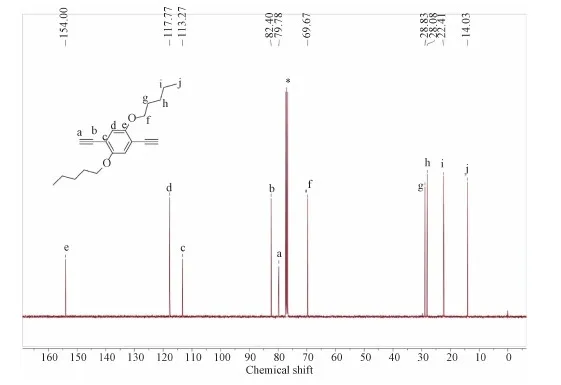
Figure S5 13C NMR spectrum of M4 in CDCl3.The solvent peaks are marked with asterisks
HRMS(EI, m/z)Calcd for[M+H]+ C20H26O2: 298.1933, Found: 298.1925, error:-2.68
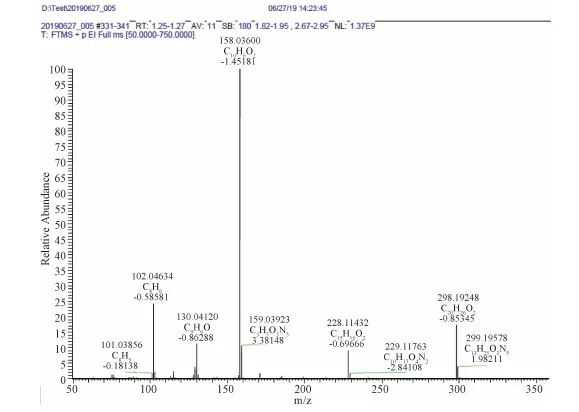
Figure S6 HR-MS spectrum of M4
2.Structural characterization of polymers
2.11H NMR spectra of polymers
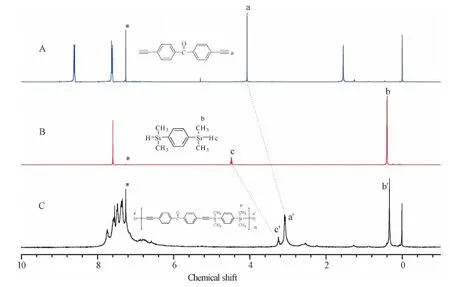
Figure S7 1H NMR spectra of(A)M2,(B)M5 and(C)P2 in CDCl3.The solvent peaks are marked with asterisks
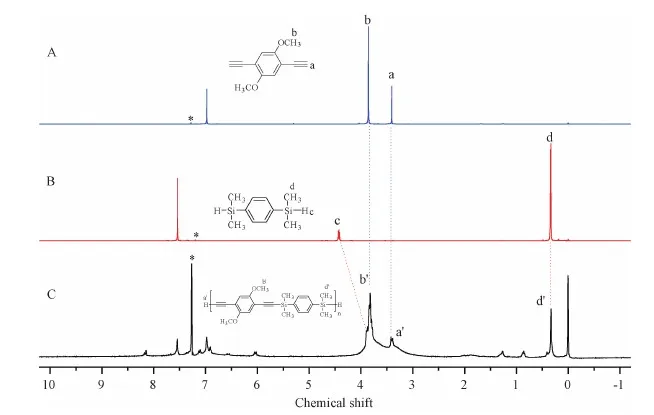
Figure S8 1H NMR spectra of(A)M3,(B)M5 and(C)P3 in CDCl3.The solvent peaks are marked with asterisks
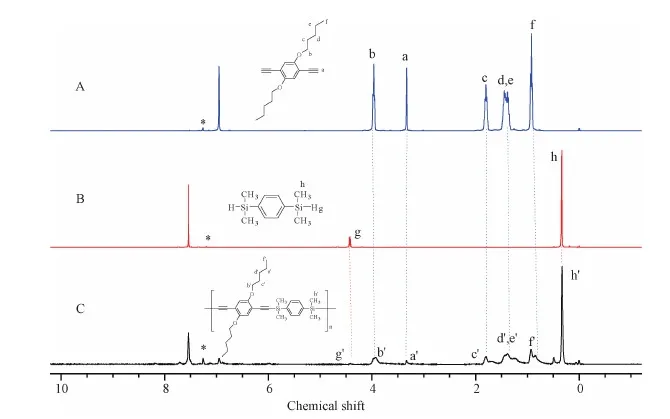
Figure S9 1H NMR spectra of(A)M4,(B)M5 and(C)P4 in CDCl3.The solvent peaks are marked with asterisks
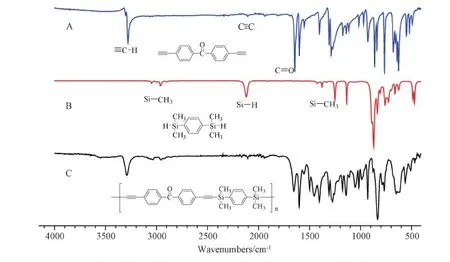
Figure S10 FT-IR spectra of(A)M2,(B)M5 and(C)P2
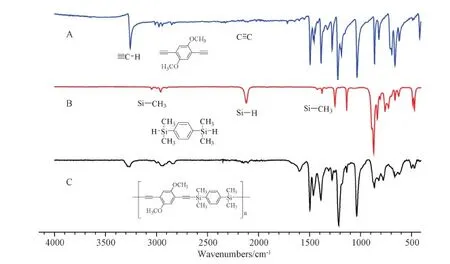
Figure S11 FT-IR spectra of(A)M3,(B)M5 and(C)P3
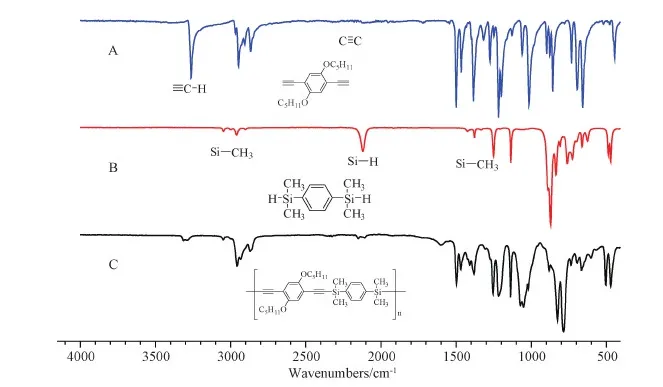
Figure S12 FT-IR spectra of(A)M4,(B)M5 and(C)P4
