
大气气溶胶中的铁及其溶解度研究综述
2021-08-25 11:21齐宇轩周杨
海洋气象学报 2021年2期
齐宇轩,周杨
(中国海洋大学海洋与大气学院,山东 青岛 266100)
引言
具有生物活性的Fe进入生物地球化学循环中能够改变固碳速率,影响海洋与大气的碳交换[1];能被海洋浮游生物利用,在光合作用、呼吸作用和固氮中发挥作用,引起浮游生物量的增加,进而影响海洋初级生产力[2-5]。研究[3]表明,该现象在高营养盐、低叶绿素(HNLC)的海域尤为明显,而HNLC海域约占世界海洋的30%。海洋中生物可利用的Fe具有丰富的来源,不仅包括大陆架沉积物的夹带与河流输入、海底热液与火山作用、冰川(冰盖)、冰山及海冰的释放,也包括大气的干湿沉降[6-8]。
大气气溶胶可通过直接散射或吸收太阳光影响大气辐射平衡,也可作为云凝结核或冰核影响云生命周期而间接影响全球气候[9-11],包括自然排放的沙尘、海盐和火山灰等,也包括人为源气溶胶[12]。沙尘传输导致的沉降是以往研究的热点[13],但近年来,更多研究者[14]开始关注复杂多样的Fe来源及其循环过程。人为排放的一次污染物会经过复杂的物理化学过程生成大气细颗粒物(空气动力学直径Dp≤2.5 μm,PM2.5),通常有更长的大气寿命,能进行远程越境或洲际传输,并向偏远地区沉积[15]。
尽管Fe在大气颗粒物质量中占比很小,但其沉降入海后能影响海洋生态系统[5],例如在我国近海,亚洲沙尘与污染气溶胶混合后,三价铁(Fe(III))可被二氧化硫(SO2)等还原成生物能直接利用的二价铁(Fe(II)),从而促进浮游植物生长[16]。目前普遍认为可溶性Fe被生物利用的概率较大[4, 17-18],国内外对大气气溶胶中Fe及其溶解度(可溶部分浓度/总浓度×100%)的研究取得了较多成果,也仍存在一些不足。
1 气溶胶中Fe的质量浓度
大量研究测定了沙尘、城市或海洋气溶胶中的总Fe质量浓度,图1总结了部分代表性站点或区域的结果[18-40],在这些观测中,全球不同地区气溶胶中的总Fe质量浓度差异很大(0.77~4 168.00 ng·m-3),最大差距接近4个数量级。受沙尘影响显著的北非附近海域以及受沙尘和人为污染共同影响的东亚及其沿海地区Fe质量浓度较高,而偏远海域如中纬度大西洋、南大洋和东太平洋等地区Fe质量浓度较低,表明Fe的源区对其全球尺度上的质量浓度分布有重要影响。对北黄海气溶胶的研究也发现近岸海域Fe质量浓度相对远海海域明显较高,说明陆源污染物对气溶胶中Fe的分布有较大影响[41],即在较小的区域尺度上也与全球的空间分布有相似特征。从粒径角度分析,同一站点的粗粒子(PM10,Dp≤10 μm的颗粒物或TSP,总悬浮颗粒物)中的Fe含量明显高于细粒子,但差距不超过1个数量级,表明细粒子中的Fe含量不容忽视。

图1 全球不同地区大气气溶胶中的Fe质量浓度分布(色标,单位:ng·m-3;图下文本框为图中位置数字对应的观测地点-观测季节-样品类型[18-40])Fig.1 Distribution of total Fe mass concentrations (colorbar, units: ng·m-3) in atmospheric aerosols in global distinct regions (The textbox below the figure shows the observation location-season-aerosol type corresponding to the position numbers in the figure [18-40])
在部分海域的研究发现由于气溶胶来源不同,总Fe质量浓度具有明显的季节变化。例如北黄海不同季节的总Fe质量浓度在326.5~2 111.0 ng·m-3,春季由于沙尘的发生[41],矿物源Fe的质量浓度最高,夏季则由于降水对颗粒物的清除作用导致Fe质量浓度最低。东海气溶胶中的Fe具有类似特征,春秋两季受沙尘影响,Fe质量浓度较高,而夏季受来自周围海域的清洁气团影响,Fe含量最低[18, 42]。
2 气溶胶中Fe的溶解度
尽管来自非洲、亚洲、南美和澳大利亚干旱地区的矿尘的大气传输和沉降是向上层海洋提供可溶Fe的主要方式[43],但近期越来越多的研究表明人为源排放的Fe溶解度更高,矿物中的不可溶Fe也能在传输过程中发生物理化学变化,从而向可溶性Fe转化。因此对大气气溶胶的研究不应局限于沙尘气溶胶,了解人为排放的细颗粒物中Fe质量浓度及溶解度也对认识Fe对海洋生态系统的影响至关重要。
以往的研究应用不同的浸出方案提取气溶胶膜样品中的可溶性Fe,得到广泛的溶解度估计值(0.1%~80.0%)[39]。通过比较大量结果,TRAPP et al.[44]和SHOLKOVITZ et al.[43]都认为控制Fe溶解度的因子很大程度上是气溶胶自身的固有特性,而与浸出方案关系不大。但总结历史数据发现,不同类型的浸出液对所得的溶解度可能有明显影响[40],例如KADKO et al.[40]分别用超纯去离子水(以下简称“纯水”)和乙酸铵溶液做介质提取可溶性Fe,所得的溶解度差别较大。关于这两种浸出方案,SHELLEY et al.[45]认为用纯水作浸出液得到的是溶解度的下限,它是保守的;而用乙酸铵作浸出液得到的是溶解度的上限,即元素潜在的最大可溶部分,两种方案相结合可以提供元素的“溶解度窗口”。海水作为另一种常用浸出液,其复杂的动力物理化学形态和微生物学在不同地区和季节之间差异很大,因此PERRON et al.[46]建议使用纯水、弱碱或强酸而非海水浸出方案,以方便进行数据间的比较。图2为部分文献中全球不同区域的Fe溶解度分布,考虑到数据需要有较好的重现性及可比性[43],在统计数据时,优先选择纯水作为浸出液所得的溶解度,其次选择乙酸铵作为浸出液所得的溶解度。
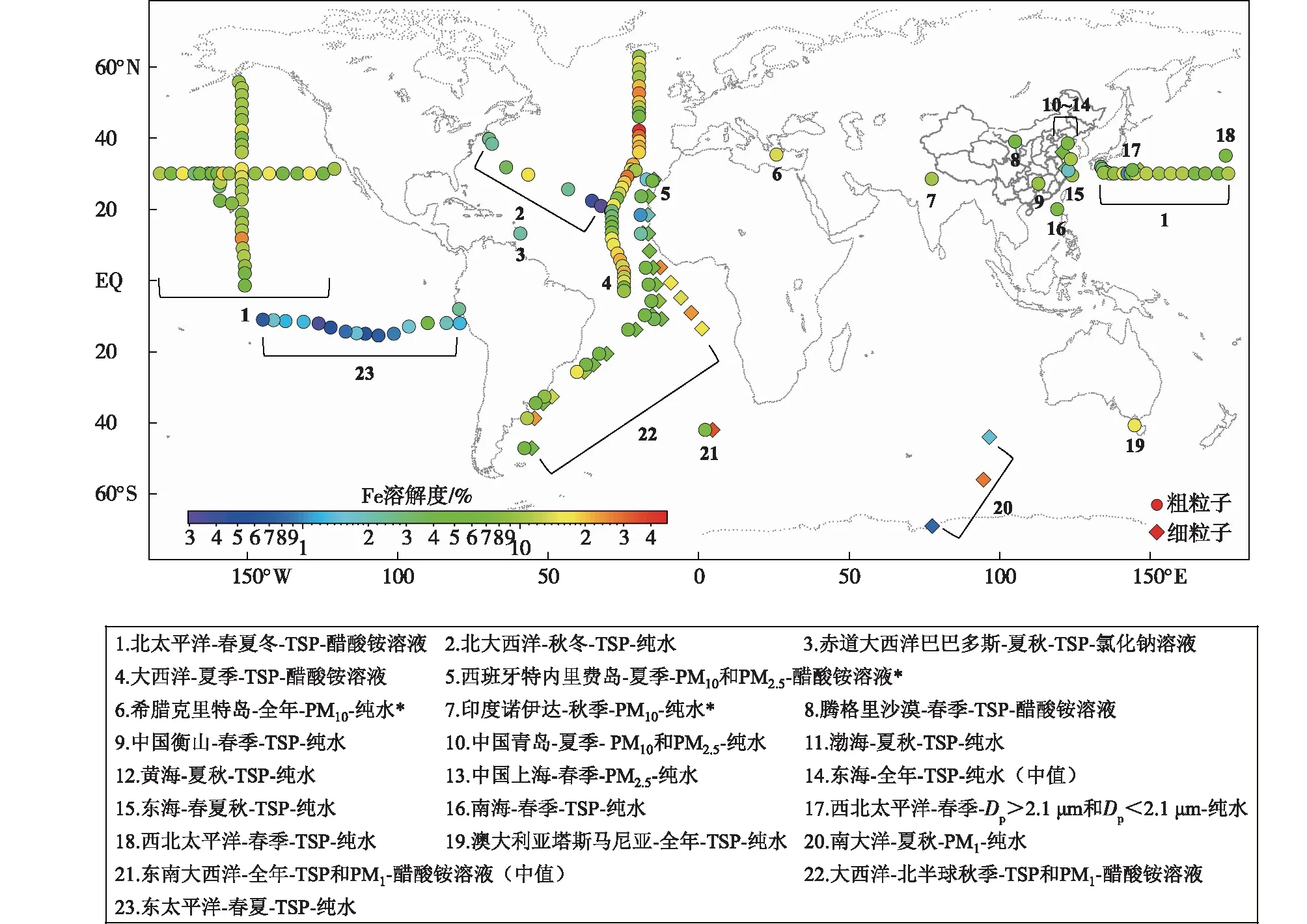
图2 全球不同地区大气气溶胶中的Fe溶解度测量值(色标,单位:%;图下文本框为图中位置数字对应的观测地点-观测季节-样品类型-浸出液[18,19,21,23,29,32,34-40,44,49-50];*溶解度为本文作者根据原文附录中的数据计算)Fig.2 Distribution of Fe solubility (colorbar, units: %) in atmospheric aerosols in global distinct regions (The textbox below the figure shows the observation location-season-aerosol type-leaching methods corresponding to the position numbers in the figure [18,19,21,23,29,32,34-40,44,49-50]. *Solubility is calculated by the author of this article based on the data from supporting information)
总体而言,Fe溶解度的全球分布与总Fe质量浓度呈完全不同的趋势。由于撒哈拉沙尘是北大西洋盆地的主要矿尘来源[47],因此北非附近的大西洋,Fe溶解度很低(0.3%~1.5%),而人为污染严重的东亚等地区Fe溶解度明显上升(1.8%~10.4%)。在东南大西洋和南大洋附近观测到的溶解度最高(47%和27%),但这些海域Fe溶解度的变化范围很大(0.3%~47.0%),与浸出方案、气溶胶的酸碱度、源区和传输路径等有关[21]。ZHUANG et al.[48]认为偏远海洋气溶胶中的Fe溶解度远高于土壤源物质中的;SHI et al.[17]和TRAPP et al.[44]发现在偏远海洋和污染区域,沙尘质量浓度较低时观测到较高的Fe溶解度。与总Fe质量浓度相反,同一站点的细颗粒中的Fe溶解度往往数倍于粗颗粒,例如,2011年西班牙特内里费地区的粗粒子中的Fe溶解度为1.54%,细粒子中的为3.29%[23];2004—2012年东南大西洋粗粒子中的Fe溶解度中值为7%,PM1中的为33%[38]。
目前对气溶胶Fe质量浓度及溶解度的研究更多地集中于粗粒子中,对细颗粒物研究较少。从沉降角度,陆源污染物中的细粒子更易长距离传输至远洋海域,且污染气团的酸性环境易使不溶Fe向可溶Fe转化。尽管在偏远地区进行巡航观测相对困难,但其对于记录和理解气溶胶的大气过程非常重要[51],可通过加强陆地站点细粒子中可溶性Fe的观测对海洋数据进行预估。
3 Fe溶解度的主要影响因素
影响气溶胶Fe溶解度的因素是多方面的,包括提取介质的pH、元素富集程度、溶液颗粒负载、气溶胶类型和粒径、光还原作用、云/老化过程、有机、酸性或碳质物质的存在及海水特性等[42, 52]。BAKER et al.[39]提出气溶胶本身的特性和来源是主要控制因素。TRAPP et al.[44]将Fe溶解度差异归因于两种假设:其一关注物理差异,Fe溶解度大部分取决于颗粒表面或附近的Fe,粒径较小的颗粒具有较高的比表面积,可提取的Fe比大颗粒多;其二关注化学差异,认为气溶胶的初始组成才是控制Fe溶解度的因素,有研究发现化石燃料和生物质燃烧产生的气溶胶中Fe溶解度较高,支持了后一种假设[39,53-55]。杜志恒等[7]将主要控制因素概括为陆地传输过程、海洋过程和人类活动三方面。
本文将专注于气溶胶沉降到海洋之前的过程,从Fe的来源、大气物理过程,以及大气化学和传输混合过程三个方面总结Fe溶解度的影响因素。
3.1 Fe的来源及气溶胶颗粒的矿物组成
Fe溶解度与气溶胶组分的来源和性质有关[44]。BAKER et al.[39]发现大西洋气溶胶中的Fe溶解度为1.4%~54.0%,随气团类型/气溶胶来源而发生明显变化,如源区为撒哈拉沙尘的Fe溶解度(中值1.7%)明显低于其他源区(中值5.2%)。DESBOEUFS et al.[56]对不同类型气溶胶的云过程模拟发现,气溶胶的来源影响微量元素的溶解度,如Fe在飞灰中的可溶性比在黄土中高950倍。
沙尘是总Fe最主要的来源,但Fe的溶解度受到其矿物学特性的影响[57-59],JOURNET et al.[58]把矿物Fe依据成键方式分为4类:具有强Fe-O(铁-氧)晶格键的由Fe、H和O组成的Fe的(氢)氧化物,如针铁矿、赤铁矿等;由共价键结合在铝硅相晶格中的Fe,如绿脱石、贝得石等;取代Mg(镁)或K(钾)形成离子键而固定在晶格中的Fe,如正长石、伊利石和蒙脱石等;无定形游离Fe,如高岭石、奥长石等。研究发现无定形Fe溶解度最高,取代碱性元素形成离子键的Fe溶解度大于在晶格中取代Si(硅)和Al(铝)的Fe溶解度,Fe氧(氢)化物溶解度最低。黏土比石英具有相对更高的Fe溶解度,在长距离传输过程中沙尘矿物会从(粒径较大的)石英到(粒径较小的)黏土的转变,造成Fe溶解度的提高[58, 60]。尽管每年沉降到海洋表面的沙尘总通量在1.490~1.814×1012t,并且沙尘中的总Fe丰度达3.5%[44, 61-63],主导了总Fe向全球海洋的输入[39],但其中只有很小一部分是易于生物利用的可溶形式[14],其溶解度通常仅在约1%或更低[43],因此Fe的其他来源,尤其含有较多可溶性Fe的来源值得重视。
除了沙尘,全球大气还有约5%的Fe来自火山爆发、生物质燃烧和人为燃烧[64],其中火山灰的表面生成过程和喷发羽流中的高低温非均相化学反应能对火山灰在大气传输期间的(光)化学反应产生影响,是火山灰Fe溶解度的控制因素[8]。WANG et al.[65]估算了1960—2007年煤炭、石油、生物燃料和生物质等燃烧源排放的Fe,发现其排放速率为0.2~9.5 ng·m-2·s-1;LIN et al.[66]的研究表明2007年燃煤飞灰排放的Fe为0.05~55.00 ng·m-2·s-1。燃烧源还是可溶Fe的重要来源,在遥远的南大洋也是如此[37,39,52,67]。ITO and FENG[68]计算了2001年东亚地区燃烧源排放的可溶Fe(0.01~0.20 ng·m-2·s-1),比LUO et al.[67]、WANG et al.[65]和CHEN et al.[69]计算的总Fe排放低近1~2个数量级。
GUIEU et al.[53]发现生物质燃烧是可溶性Fe向开放海洋传输的重要来源,其溶解度为2%。尽管从全球Fe收支来看生物质燃烧输入的Fe仅为沙尘输入的10%,但其能对局地造成显著影响。BOWIE et al.[6]发现亚南极地区的矿尘气溶胶中Fe溶解度在0.2%~2.5%,但生物质燃烧排放的颗粒中溶解度为17.7%。TRAPP et al.[44]测量出Fe溶解度最高(5.8%)的样品受非洲南部生物质燃烧的影响。ITO[70]通过模式估算发现来自生物质燃烧源的Fe溶解速率较快,对东南亚、非洲南部、澳大利亚和南美地区的热带和南部海洋下风处的可溶Fe贡献显著(40%~60%)。然而,生物质燃烧气溶胶中Fe溶解度的范围很大,从0.5%~46.0%变化了2个数量级[53, 70-72],ITO[70]认为可能与硫酸铁、无定形铁和稳定的铁有机络合物有关[72-74],并通过优化全球气溶胶化学传输模式(IMPACT)证明森林火灾对可利用Fe的贡献很大,缺失源(如煤矿开采活动、火山爆发和二次生成)也有重要贡献[75]。
来自人为燃烧气溶胶的可溶Fe通量在向表层海洋输入的总可溶Fe中占比也较大,特别是在远离主要土壤尘源区的海域,如南大洋和南太平洋[76]。人为燃烧中燃油源Fe的溶解度(36%~81%)又明显高于燃煤源(0.2%~25.0%),尽管航运对Fe的贡献相对较小(约2%),但航运产生的Fe主要沉降在附近海洋中,是北半球偏远地区和赤道太平洋HNLC海区可溶性Fe的重要来源(达50%)[52, 70, 77-78]。模式结果也显示,虽然人为源Fe占全球总气溶胶Fe的比例小于5%,但在某些区域可能占到Fe沉降的50%[67]。与矿尘气溶胶相比,来自燃烧源的Fe更可溶,也更易在较细颗粒中发现[79],因此细粒子中的Fe及其溶解度需要更多关注。
3.2 大气物理过程
Fe的不同来源意味着含Fe颗粒物的粒径不同,在重力作用下产生不同的沉降速率,较大的颗粒优先沉降去除。已有研究[80]证明Fe溶解度是颗粒物粒径的函数,传输过程中沙尘颗粒浓度随行进时间或距离呈指数下降[81],颗粒平均模态粒径逐渐减小[54],该现象主要发生在距离源的前1 000 km内[82]。因而当气团传输到开阔大洋时,沙尘浓度降低,沙尘颗粒的比表面积增加,使更多Fe位于接近颗粒表面的位置,易被提取而溶解[44]。
BAKER and CROOT[83]发现沙尘浓度和Fe溶解度呈反比关系,可验证上述假说。SHOLKOVITZ et al.[43]归纳了上千个气溶胶样品数据,发现Fe溶解度随总Fe负荷的变化在全球呈现基本一致的双曲线趋势。但粒径大小并非造成这种反比关系的唯一原因,因为矿物气溶胶浓度的降低多为大气传输的作用,所以传输中的其他过程也可能导致这种Fe溶解度趋势[83],如较细的沙尘颗粒(≤3 μm)中可溶性Fe质量浓度的增加可能是大气传输过程中与酸性硫酸盐气溶胶相互作用的结果[84]。MAHOWALD et al.[52]用一个简单一维羽流模型说明了这种反比关系是由于不同来源和大气过程造成的。事实上,Fe溶解度的变化可能是大气物理过程、大气化学过程与传输混合过程综合作用的结果[85]。
3.3 大气化学与传输混合过程
气溶胶传输过程中还会发生大气化学与传输混合过程。HSU et al.[42]发现沙尘粒径的减小导致有更多的表面积作为酸性成分的汇,导致Fe等沙尘源元素溶解度增加,表明大气物理和化学过程相互关联。但大气化学过程增加Fe溶解度的反应机理尚未达成共识。一种可能是发生了Fe的还原反应,Fe(II)高度可溶,Fe溶解度的主要变化受来自人为源的Fe(II)驱动;Fe(III)的溶解度更稳定,受人为源和浸出方法的影响较小,导致的Fe溶解度变化也较小[44],而在大气水的凝结和蒸发循环期间,酸性无机和有机物种(尤其是人为的SO2、NOx(氮氧化物)及其氧化产物)和/或光照可促进Fe(III)向Fe(II)转化[43, 83, 86-88]。另一种可能是发生了非还原性溶解过程[89],如酸过程和云过程都可通过生成溶解形式的Fe(III)而使Fe溶解度增加。本文将主要化学过程归纳为:光化学过程、酸过程和云过程。
3.3.1 光化学过程
20世纪有研究[90]表明,日光照射和草酸盐的添加有利于海洋气溶胶Fe(II) 质量浓度增加,尽管该研究认为光致还原作用并未明显促进沙尘矿物基质中Fe(III)的溶解,但CHEN and SIEFERT[91]的实验对比了添加化学还原剂与太阳光照射对Fe溶解的影响,证明光致还原作用可以明显促进Fe溶解。相对难溶的Fe(III)转化为更易溶的Fe(II),而草酸盐等有机物会促进这种还原反应[89]。BAKER and CROOT[83]也认为有机配体如甲酸盐、乙酸盐和草酸盐与Fe(III)可以在颗粒表面形成络合物,作为电子供体,通过光还原促进Fe(II)生成并释放到溶液中。草酸盐等有机酸能充当还原剂和“桥梁”,既为颗粒物上的Fe(III)转移电子,又与新生成的Fe(II)络合,使其从颗粒物表面释放到溶液中[92-93]。CHEN and GRASSIAN[94]对沙尘和燃煤颗粒中的Fe用不同种类的酸进行浸提,发现草酸盐浸提效率最高,光照会促进Fe的溶解。
Fe(III)-草酸盐的光降解机制的反应过程概括如下:[Fe(III)(C2O4)3]3-的初始路径有两种方式,通过Fe(III)-O键的断裂(反应(1a)、(1b))[59, 95-96],或者发生分子内电子的转移生成C2O4·-(反应(2a))并发生C2O4·-的降解(反应(2b))生成二氧化碳(CO2)[97-100]。
(1a)

(1b)
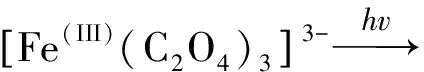
(2a)

(2b)
两种路径都会生成具有较强还原性的Fe(II)和CO2·-, 同另一个[Fe(III)(C2O4)3]3-发生还原反应(3),将氧气(O2)降解为O2·-(反应(4))。
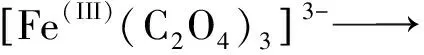
(3)
(4)
生成的O2·-可同氢离子(H+)反应生成HO2·(反应(5)),HO2·在酸性环境下发生自反应生成过氧化氢(H2O2)(反应(6)),H2O2又会氧化Fe(II)生成·OH(反应(7)), 完成这个氧化还原循环过程。
O2·-+H+↔HO2·
(5)
2HO2·↔H2O2+O2
(6)
Fe(II)+H2O2↔Fe(III)+·OH+OH-
(7)
该过程不仅影响草酸盐丰度,同时生成了大量H2O2、O2、·O2、·OH和HO2·等活性氧自由基(ROS),可以改变大气氧化性从而影响大气化学过程。目前很多大气模式未加入该过程,导致高估草酸盐的丰度[101-102],但基于对该过程的实验室模拟构建的模式,如CAPRAM 3.0i(液相化学自由基机制)等,又明显高估该过程的影响[103],表明实验室模拟和真实大气环境状况存在差别。单颗粒质谱仪的引入可从单个颗粒物的角度分析含Fe颗粒物的混合状态,ZHOU et al.[104]发现含草酸盐和Fe的颗粒物浓度在日出后明显降低,表明二者络合物的光解,光化学过程生成大量ROS促进单个颗粒上硫酸盐的生成。进一步发现,人为源尤其是工业源排放的草酸盐-Fe颗粒物的降解速率最高,沙尘和生物质燃烧源次之,垃圾焚烧源排放的含草酸盐-Fe颗粒物几乎没有光学活性。生物质燃烧排放的草酸盐相当一部分是通过二次氧化生成的,ROS促进硫酸盐生成的同时也促进草酸生成并提高Fe溶解度,因此,生物质燃烧源Fe的溶解度虽较高,但其含草酸盐Fe颗粒物的降解速率并非最高。这些研究表明真实大气环境的颗粒物混合状态复杂多变,也会对其光化学过程产生影响。
3.3.2 酸过程
有研究[56]提出,液相pH能控制气溶胶溶解度;酸化过程是控制微量元素溶解度变化的主因[105-106];LI et al.[107]观测到沙尘期间微量元素溶解度明显下降,提出碱性物质导致气溶胶酸度降低,抑制元素溶解。酸溶解过程往往伴随着传输混合过程,强调自然和人为排放的无机物、有机物及内外混合作用对溶解度的影响[7]。矿物气溶胶经过人为污染严重的区域时,能与SO2、NOx等无机物种混合并发生非均相反应,颗粒物表面溶液变成强酸性,高浓度的质子(H+)破坏铁氧化物(如Fe2O3,氧化铁)中的Fe-O键的稳定性,从而促进低pH条件下Fe的溶解[79, 89, 108]。生成的硝酸盐和硫酸盐吸湿性较强[109-111];气溶胶传输至海洋上空时与海盐气溶胶混合,与Cl-(氯离子)发生非均相反应生成CaCl2(氯化钙),同样具有较强的吸湿性[111-114],进而增大颗粒表面的反应面积,促进Fe溶解。单位体积空气中颗粒物的表面积越大,酸性表面反应造成的Fe的溶解效率就越高[115]。
BAKER and CROOT[83]认为在酸增强Fe溶解度的背景下,气溶胶酸性组分和矿尘组分的内混是必要的先决条件,酸增强溶解度的速率取决于沙尘和(游离)酸性物种的内混速率。碱性沙尘与含铁矿物质的混合状态的变化可能导致较小的沙尘颗粒产生更多的可溶Fe,该混合状态是粒径的非线性函数[79]。此外,酸化过程中的有机络合能显著提高Fe溶解度[77],比起硫酸和乙酸,草酸能更大程度上促进Fe溶解[94],其在含Fe颗粒物表面形成有机配位体,使Fe-O变得不稳定以至于断裂,产生溶解形式的Fe(III)来增加Fe溶解度[57, 89]。PARIS et al.[116]的浸出实验显示,pH值=4.7时,草酸可使非洲沙尘中的Fe溶解度从0.003%增至0.250%。LUO and GAO[117]的模式研究表明,草酸可使沙尘中的Fe溶解度增加1%~3%。JOHNSON and MESKHIDZE[118]的模式结果显示,与仅考虑质子促进Fe溶解的情况相比,在模式中加入草酸盐和芬顿化学反应使预测的全球海洋可溶Fe的年沉降量增加了约75%,但该模式未考虑Fe化学在草酸盐形成中的作用。ITO[70]的模式模拟也得出类似的结论,草酸盐的添加使从燃烧源沉降到全球海洋中的可溶Fe增加了1倍以上。
酸过程对可溶性Fe增加的作用和机制已有许多实验室和模式模拟,但很少观测到酸浓度(如硫酸盐)和Fe溶解度间的相关[52]。KUMAR et al.[119]和SRINIVAS et al.[120]基于Fe溶解度和非海盐硫酸盐之间的显著相关关系,认为酸溶解能使气溶胶Fe溶解度增加,但非海盐硫酸盐浓度高的气溶胶可能与燃烧排放有关,而这些燃烧气溶胶固有的Fe溶解度本身就高于沙尘,所以这种相关性并不意味着酸和Fe溶解度间一定存在机制上的关联[43]。不过最近LI et al.[121]的单颗粒分析发现,富Fe颗粒在大气中停留1~2 d后会覆盖酸性的厚硫酸盐层,Fe以可溶形式存在于其中,随老化程度(涂层厚度)增加而增加,证明酸性条件确能促进Fe溶解。
3.3.3 云过程
3.4 各影响因素间的相对重要性
基于对Fe溶解度控制因子的认识,许多研究探讨了各因子的相对重要性。总体而言,无法确定某个单个过程可作为Fe溶解度的主导控制因子,根据地区和季节的不同,各因素的协同效应更为重要[52]。BAKER et al.[39]的实验室研究表明,颗粒物浓度效应对Fe溶解度的影响较小,酸过程可以提高Fe溶解度,但该过程的重要性无法确定。HSU et al.[42]认为各控制因子的重要性排序可能为:酸过程(大气过程)>化学形态≈初始组分(来源)>沙尘负载,并且大气过程对溶解的影响对于地壳元素比人为元素更重要。SHI et al.[85]通过实验室和模式研究证明,重力沉降虽能导致沙尘中Fe溶解度增加,但其增加比起在热带和亚热带大西洋观测到的低1个数量级,由此认为一旦撒哈拉沙尘到达大西洋, Fe溶解度增加的主要机制就不再是物理的重力沉降,而是颗粒物的化学反应和/或与人为气溶胶的混合等大气过程。SHOLKOVITZ et al.[43]总结,大气化学过程造成的土壤尘Fe溶解度的增幅多在1%~5%,而全球数据显示大部分Fe溶解度>10%,由此认为大气化学过程和气溶胶来源(尤其是燃烧气溶胶的排放)都是导致低气溶胶Fe负载下溶解度升高的原因,二者的相对重要性因区域而异,但在全球尺度上后者最为重要。因此,在分析Fe溶解控制因子间的相对重要性时,要综合考虑季节、地区和空间尺度等要素,并将实验室和模式模拟数据与实测数据进行对比分析。
4 结论
根据上述讨论,我们有以下发现:
1)Fe溶解度各控制因子的相对重要性是可变的[42],可能和季节、距气溶胶源地的距离、研究区域、空间尺度或其他因素有关,研究时要具体情况具体分析。
2)Fe溶解度各控制因子之间相互关联。颗粒物发生重力沉降的速率与其来源和矿物组成有关;云过程和酸过程可以促进非光化学反应中的非均相反应;云过程和大气物理过程都可以促进酸过程;大气物理过程和气溶胶的老化、云和酸过程等大气化学过程都与传输混合有关。可见这些过程都不能轻易与彼此区分开[42, 83]。
未来关于气溶胶Fe的研究可以更加关注以下方面:
1)关注海洋气溶胶Fe及其他微量元素,包括总质量浓度、可溶部分质量浓度及溶解度的控制因素,建立更多的长期观测来研究气溶胶Fe及其对海洋的影响[52]。
2)细模态中含离子的矿物形态对于研究不同粒径颗粒的Fe溶解度很重要,目前关于矿尘Fe对海洋生产力的作用的研究多关注较大的颗粒[79],也需要更加关注陆源或细颗粒物中的Fe以及海洋气溶胶中由船只排放或石油开采等活动排放的Fe。
3)应结合粒径分级矿物学,研究不同粒径模态的气溶胶中Fe的大气过程[79]。
4)基于全样气溶胶的分析无法将不同来源的含Fe颗粒物区分开,而含Fe气溶胶发生的物理化学过程可能由于单颗粒混合状态的不同而有所区别,需将其混合状态与可溶Fe的分布及其与酸性化合物的关系联系起来[121],将单颗粒分析与全样分析相结合开展更为综合的观测研究。
5)将外场观测、实验室研究和模式模拟等多种研究手段结合,包括三维建模和综合工作[52],注重源清单的建立,在模式中应全面考虑热源/燃烧源和成岩源/沙尘源的含Fe气溶胶[127],以准确预测生物可利用Fe的沉降通量并对各种Fe溶解度影响因素的作用进行量化。
应指出,本文讨论的Fe溶解度多数是实验室分析的估算值,关注气溶胶沉降至海洋前的过程。但实际上,气溶胶沉降在海洋表面后,所含Fe和其他微量元素的溶解度和生物可利用度还受海洋过程的影响,包括多种物理、化学及生物因素,如海水pH、停留时间、有机配体、光化学作用、超氧化、掠食和吞噬作用等[70, 83, 128-129]。因此,需综合考虑气溶胶本身性质、大气过程及海洋过程,将多学科、多种研究方法和手段结合,以深入了解Fe的整个生物地球化学循环过程及其影响[7]。
猜你喜欢
成都信息工程大学学报(2022年3期)2022-07-21
地球科学与环境学报(2021年6期)2021-11-16
世界科学技术-中医药现代化(2021年5期)2021-11-05
装备环境工程(2019年6期)2019-07-16
试题与研究·中考化学(2016年4期)2017-03-28
中学化学(2016年10期)2017-01-07
中学化学(2015年5期)2015-07-13
国外科技新书评介(2014年4期)2014-12-17
